
系统性归纳鼻腔鼻窦小圆细胞肿瘤(small round cell tumors,SRCT)的临床病理、免疫组织化学特征及分子遗传学进展,重点关注诊断及新进展,为临床治疗及预后判断提供依据。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
小圆细胞肿瘤(SRCT)一般指病理学上表现为小细胞、核深染、核质比高的一组肿瘤实体,几乎都是恶性肿瘤,极少数为良性。SRCT临床及影像学有相似性,病理学上具有重叠,另外由于取材小、破碎、手术钳夹伤等原因诊断困难,我们结合工作经验、复习文献,探讨该组肿瘤临床病理学特征,以期对该组肿瘤精准诊断,指导临床治疗并为预后提供依据。
(1)临床:肿瘤起源于嗅神经上皮,占鼻腔肿瘤5%,男性略多见(男女比为1.9∶1.0),年龄3~90岁,高发于20~30岁及50~60岁两个年龄段[1]。早期无明显症状,后期可表现为鼻塞、鼻出血、嗅觉下降、头痛、突眼、视力模糊等。CT检查常为实性软组织肿块密度影,可侵犯周围组织,增强后不均匀强化。(2)病理学:低倍镜下呈结节状、分叶状结构;瘤细胞片状、实性、巢状、梁状、簇状排列,形成"器官样结构"(图1A),可见多种类型的菊形团,如Homer-Wright菊团(中央为神经基质),Flexner-Wintersteiner型菊形团(中空管腔),可有神经毡岛;瘤细胞圆形、卵圆形,染色质细腻,间质富于毛细血管网。少数可钙化、伴黑色素细胞分化及异源性分化(腺样、鳞化、畸胎瘤及横纹肌母细胞分化)。罕见病例中可见肿瘤于黏膜上皮内生长,甚至肿瘤局限于黏膜上皮内,上皮可鳞化或增生,属于原位ONB[1](图1B)。肿瘤分四级:Ⅰ级,细胞温和,无核分裂象和坏死,常见Homer-Wright菊形团及神经毡岛,可钙化;Ⅱ级,细胞轻度异型性,核分裂象少见;Ⅲ级,细胞异型性明显,可见核分裂象及坏死,可见Flexner-Wintersteiner菊形团;Ⅳ级,奇异型细胞,核分裂象及坏死明显。(3)免疫组织化学:瘤细胞神经元特异性烯醇化酶(NSE,图1C)、突触素、嗜铬粒素(Cg)A、CD56、Calretinin阳性,S-100蛋白染色可显示支持细胞(图1D),部分CK阳性,Calretinin阳性可与其他小圆细胞肿瘤鉴别,PHOX2B是神经母细胞瘤一个相对特异性的标志物,有助鉴别诊断[2]。CD34、CD99、HMB45、Melan A、MyoD1、Myogenin、结蛋白和h-caldesmon阴性。(4)遗传学:染色体8q的获得或缺失,无EWSR1基因重排及MYC基因扩增。(5)电镜:胞质内见神经内分泌颗粒、神经细丝、微管和微丝及神经突样。(6)治疗及预后:手术、放化疗等。预后与肿瘤临床分期、病理分级、手术是否完整切除及是否联合治疗等有关,总体10年生存率在70%左右。

(1)临床:好发儿童及青少年,男性略多见,临床表现有鼻腔异物感、鼻堵、鼻腔肿物等。CT检测为软组织肿块影,病变较小时不累及鼻骨及其邻近结构。(2)病理学:弥漫性一致小圆细胞呈实性、片状、巢状分布,瘤细胞圆形、卵圆形(图2A),部分短梭形,染色质细腻,胞质淡染或透明(含糖原),瘤细胞常温和,一般无瘤巨细胞,核分裂象不等(图2B),部分可见坏死;间质见丰富毛细血管网。"Ewing肉瘤样肿瘤"指在形态学与EWS/PNET相似,但免疫表型、遗传学不同的异质性肿瘤实体,主要包括CIC基因重排的肉瘤、BCOR-CCNB3基因易位的肉瘤、BCOR末端重复序列相关性肉瘤(ITD)等[3]。(3)免疫组织化学:NSE、突触素、CgA、CD56阳性,其次CD99膜阳性(图2C)、Fli-1、NKX2.2阳性,少数ERG阳性。CIC基因的重排的肉瘤ETV4和WT1阳性,而FLI1、NKX2.2、ERG阴性;BCOR-CCNB3基因易位的肉瘤呈BCOR阳性,而ETV4、NKX2.2阴性[3]。BCOR属于一种细胞核转录因子,与高级别子宫内膜间质肉瘤、肾透明细胞肉瘤等有关,BCOR阳性也见于滑膜肉瘤,偶见于横纹肌肉瘤。(4)遗传学:95%以上肿瘤具有EWSR1基因重排,90%为t(11;22)(q24;q12),形成EWSR1-Fli-1(图2D),其次为t(21;22)(q22;q12)形成EWS-ERG、t(7;22)(p22;q12)形成EWS-ETV1、t(17;22)(q12;q12)形成EWS-E1AF、t(2;22)(q33;q12)形成EWS-FEV[4]。CIC基因重排肉瘤多为CIC-DUX4、CIC-ETV4及CIC-FOXO4等;BCOR基因相关性肉瘤主要有BCOR-MAML3及ZC3H7B-BCOR融合基因类型等[5]。(5)电镜:细胞器稀少,可见高尔基体、粗面内质网、核糖体、糖原及神经内分泌颗粒。(6)治疗及预后:手术完全切除、放化疗。预后不良,少数复发及转移。
(1)临床:90%在1岁以下,女性较男性多见,90%发生于上颌骨、下颌骨、颅骨,其次为附睾、颅内、眼眶、纵隔、皮肤软组织及骨等[6]。临床表现为迅速增大的色素性肿物,严重者面部畸形。影像学显示溶骨性破坏,累及周围组织。(2)病理学:肿瘤呈簇状、巢状、片状或条索状排列(图3A);肿瘤有3种细胞,上皮样细胞、小圆形神经母细胞样细胞及梭形细胞;上皮样细胞体积较大,含有多少不等的黑色素,小细胞类似于神经母细胞,未见黑色素(图3B);梭形细胞束状排列,细胞温和,胞质淡染,部分细胞和上皮样细胞过渡移行(图3C);核分裂象罕见,无坏死。(3)免疫组织化学:上皮样细胞及梭形细胞表达CKpan、上皮细胞膜抗原(EMA)、HMB45、Melan A、NSE;小圆细胞表达NSE、突触素(图3D),部分可表达CgA、GFAP和S-100蛋白。(4)遗传学:部分有CDKN2A胚系突变及BRAF V600E基因突变。无EWSR1基因重排及MYC基因扩增。(5)电镜:上皮样细胞胞质内见桥粒、突起、基底板及不同发育阶段的黑色素颗粒;小细胞胞质内见神经内分泌颗粒、细胞突起等。(6)治疗及预后:手术完全切除;良性肿瘤,预后较好,10%~50%病例可复发,与切除不完全有关,复发多见于颌骨及颅脑,恶变、转移及死亡多见于颅脑及附睾,6%可转移到淋巴结、肝、骨、肾上腺和软组织等[6]。CD99阳性与肿瘤侵袭性相关,Ki-67阳性指数超过15%,提示高的侵袭性潜能。
(1)临床:原发于头颈部RMS占38%,多见于口腔、咽、面部和颈部软组织,一组777例RMS研究,鼻腔鼻窦RMS约占8.1%[7],分类有胚胎型(embryonal rhabdomyosarcoma, ERMS,含葡萄簇型)、腺泡状(alveolar rhabdomyosarcoma, ARMS)、梭形细胞/硬化型(spindle cell/sclerosing rhabdomyosarcoma, SC-RMS)、多形性横纹肌肉瘤及上皮样横纹肌肉瘤。鼻部以ERMS常见,其次为ARMS。年龄14~74岁(平均40.1岁),男性略多。临床表现鼻扭曲、异物感、鼻出血等,侵犯邻近组织时有面部畸形、突眼等。CT检查早期鼻黏膜增厚,可呈息肉样,呈软组织肿块密度影,随着肿瘤进展,可侵犯邻近组织。(2)病理学:①ERMS:细胞呈弥漫性、片状、实性、巢状排列,瘤细胞圆形、卵圆形、短梭形,可见横纹肌母细胞,细胞常丰富,核分裂象易见。葡萄簇型细胞密度低,间质水肿及黏液变性(图4A);②ARMS:瘤细胞实性、片状及假腺样排列(图4B),部分胞质透明;③SC/SRMS:梭形细胞为主者,瘤细胞束状、鱼骨样排列;硬化为主时瘤细胞短梭形、卵圆形为主,间质硬化及玻璃样变性(图4C);④上皮样型:细胞上皮样,泡状核,核仁明显,常见坏死、核分裂象;⑤多形性型,表现为多形性,瘤巨细胞、核分裂象易见,胞质嗜酸性,可见横纹肌母细胞。(3)免疫组织化学:MyoD1、Myogenin(图4D)、结蛋白及Myosin等阳性,异常表达如CKpan、CD34、WT1、S-100蛋白、神经丝蛋白(NF)、突触素、NSE、CD56等,少数腺泡状横纹肌表达间变性淋巴瘤激酶(ALK)和PAX5。(4)遗传学:11p15区等位基因丢失,部分有染色体2、8和13多倍体。ARMS可有t(2;13)(q35;q14)、t(1;13)(q36;q14),产生PAX3-FOXO1、PAX3-FKHR、PAX7-FOXO1融合基因;30%有12q13-15(含GL、CDK4和MDM2)和2p24(MYC)基因扩增;SC/SRMS伴有MyoD1、NCOA2基因突变,MDM2/HMGA2基因扩增[8]。(5)电镜:可见肌丝及Z带,无黑色素颗粒及神经内分泌颗粒。(6)治疗及预后:手术完全切除,术后辅以放化疗。ERMS较好,ARMS次之,SC-RMS的治疗同ERMS,预后较ERMS稍好,多形性横纹肌肉瘤预后最差。新近一组ARMS研究显示,18例无病生存(18/52,平均69.6个月),7例带瘤存活(7/52,其中6例转移,平均11个月),26例死于疾病(26/52,平均18.5个月)[9]。

(1)临床:好发年龄为儿童及老年人,男性略多,部位为颌骨、鼻骨等。影像学检查可见明确骨质破坏,侵犯骨质及周围组织(图5A),少数患者有复发病史[10]。(2)病理学:特征性小圆细胞及其直接产生肿瘤性骨样组织(图5B)弥漫性片状分布,细胞核深染,核质比高,核分裂象易见,无瘤巨细胞(图5C、图5D),可见不规则淡染的骨样基质,肿瘤呈侵袭性生长而破坏宿主骨。(3)免疫组织化学:SATB2、CD56阳性,SATB2是骨母细胞成熟转录因子,骨母细胞及其肿瘤阳性,但不能用于鉴别骨母细胞肿瘤良恶性,SATB2阳性还见于个别BCOR-CCNB3基因融合肉瘤;一般CKpan、Myogenin、MyoD1、结蛋白、S-100蛋白、SOX10、HMB45、Melan A、突触素及CgA阴性。(4)遗传学:一般无CIC及BCOR基因重排及MDM2基因扩增,极少数具有EWSR1-CREB3L1融合基因。(5)电镜:瘤细胞黏附性差,核不规则,染色质不成熟,有胶原纤维。(6)治疗及预后:手术完全切除,放化疗。预后较好,可能是肿瘤易于产生症状,利于早期发现。
(1)临床:好发于成人,年龄11~83岁(平均34.8岁,中位26.5岁),女性略多(男女比1︰2)[11]。好发部位颅底、鼻骨、鼻腔鼻窦、上颌窦、筛窦、蝶窦、口腔等,罕见颌骨。临床表现有鼻塞、鼻出血、肿物、疼痛、鼻分泌物、头痛和面部不对称。影像学检查见结节状肿物影,内见软骨样基质,可见斑点、环状钙化,肿瘤侵犯周围组织(图6A)。(2)病理学:低倍镜呈结节状、分叶状结构,可见纤维分隔带(图6B);瘤细胞主要为弥漫性一致的小圆细胞片状、实性分布,形成血管外皮瘤样结构(图6C);瘤细胞圆形、卵圆形、短梭形,核质比高,核深染,染色质细腻,散在破骨样多核巨细胞,核分裂象罕见(多数<2/10 HPF);可见分化好的软骨岛,提示软骨分化(图6D),可钙化或骨化。(3)免疫组织化学:瘤细胞呈S-100蛋白、SOX9阳性,部分CD99和NSE阳性,其他多阴性。(4)遗传学:半数普通型软骨肉瘤有IDH1/2基因突变,但罕见于MC;25%~70%的病例可检测到HEY1(8q21)-NOCA2(8q13)融合基因,少数有t(1;5)(q42;q32)产生IRF2BP2-CDX1融合基因,NCOA2、CDX基因突变与多种血液肿瘤相关,如粒细胞白血病中检测到ETV6-CDX2融合基因。(5)电镜:无基底膜样物,无微丝、黑色素颗粒及神经内分泌颗粒。(6)治疗及预后:手术完全切除为主,其次可放化疗(如环磷酰胺,甲氨蝶呤等)。肿瘤易复发,部分有致死风险,复发比例6/13,致死比例5/13,无病生存比例6/13,其中1例复发2次,术后23.8年死亡[11]。
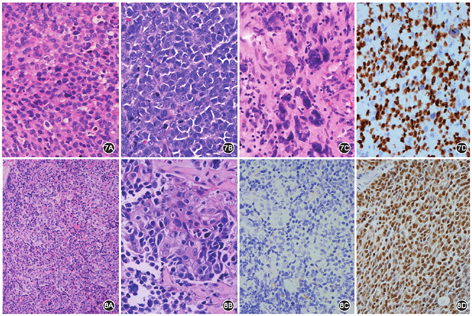
MM):(1)临床:鼻腔MM约占人体1%,好发于老年人,年龄30~90岁,男女比例相当[12]。临床表现有鼻腔异物感、流鼻血、鼻堵或无明显症状。CT检查示鼻中隔、鼻黏膜增厚,或鼻腔异常软组织密度影,增强扫描后强化不明显。(2)病理学:低倍镜下肿瘤细胞弥漫性、片状、实性、巢状、簇状、束状排列等,可破坏黏膜;瘤细胞可呈小细胞(图7A)、大细胞样(图7B)、上皮样、梭形细胞、浆细胞样及牙瓣样,罕见病例瘤细胞多形性(图7C),可见大核仁,可有核内包涵体,诊断线索是黑色素颗粒;可见核分裂象及坏死,少数间质黏液变性。(3)免疫组织化学:瘤细胞呈S-100蛋白、SOX10(图7D)及黑色素标志物阳性;异常表达有CKpan、CEA、NSE、CD117、CD99、突触素、CD56。(4)遗传学:CKIT基因突变率比例2/5,BRAF V600E突变比例1/17,NRAS突变率14.3%(8/56)~3/5[12]。(5)电镜:可见不同发育阶段的黑色素颗粒及前体物质。(6)治疗及预后:手术完全切除、放化疗及免疫治疗等,化疗药物有替莫唑胺、洛铂、顺铂及干扰素等。涉及BRAF V600E及CKIT基因突变,可选择靶向治疗如维罗非尼及甲磺酸伊马替尼等,尤其是经过维罗非尼治疗后80%患者可缓解[12]。
(1)临床:鼻腔鼻窦小细胞癌罕见,目前英文文献报道约100多例,男多于女性(男女比例69︰56),发生于成人,多见于16~89岁(平均56岁),临床上呈侵袭性进展过程,70%的患者发现时处于Ⅳ期[13]。CT检查示,肿瘤侵犯鼻黏膜、鼻甲及周围组织。临床表现有流鼻涕、鼻塞、鼻出血,后期突眼、面部变形等。(2)病理学:低倍镜下见弥漫性一致的小圆细胞浸润,片状、实性及巢状排列,菊形团结构不明显;高倍镜下瘤细胞核质比高,染色质细腻,类似于"胡椒粉样",细胞核呈卵圆形、圆形或梭形,多无核仁,部分可见小核仁,核分裂象易见(约10~30/10 HPF),细胞凋亡活跃,可有坏死;DNA裂解在血管壁周残留嗜碱性物质是线索;肿瘤间质常见丰富的毛细血管网。(3)免疫组织化学:肿瘤细胞CKpan、EMA、突触素、CgA、CD56、NSE及甲状腺转录因子(TTF)1阳性,Ki-67阳性指数较高,一般大于40%;CK5/6、p40、p63、CEA及S-100蛋白阴性。(4)遗传学:MEN1、p53基因突变及RB1基因缺失等。(5)电镜:胞质内见神经内分泌颗粒。(6)治疗及预后:主要为手术切除、放化疗,总体生存率22%(14%~30%),局部复发率45%,远处转移率35%[13],如淋巴结、肺、肝、骨髓和脊髓转移。
(1)临床:常见恶性肿瘤,年龄12~88岁(平均53岁),男性略多(男女比239︰220),临床进展较快,80.6%发病时已是Ⅳ期。影像学检查与小细胞癌类似[13]。SMARCA4(BRG1)或SMARCB1(SNF5/INI1)缺失的未分化癌属于一个罕见特殊亚型,临床呈进行性发展。NUT癌指NUT基因重排的中线癌或伴t(15;19)(q13;p13.1)易位的癌,成人好发(中位年龄30岁),男女比例相当[14],纵隔以上好发,如胸腔、纵隔、鼻、会厌、涎腺、膀胱、髂骨等。(2)病理学:低倍镜下瘤细胞呈弥漫性、片状、巢状分布,瘤细胞上皮样细胞、小圆细胞,部分泡状核,可见核仁,核分裂象易见(图8A),可见坏死,间质可有淋巴细胞及浆细胞浸润,部分类似于淋巴上皮瘤样癌表现,瘤细胞无明显的分化方向,一般无细胞间桥及角化珠,亦无腺癌成分。NUT癌表现为小圆细胞、泡状核细胞,诊断特征为瘤细胞出现骤然或陡然的角化(图8B),即瘤细胞由分化相对幼稚或不成熟的细胞突然过渡为角化,形成角化珠甚至胸腺小体样结构。(3)免疫组织化学:未分化癌CKpan、p63、p40阳性;部分未分化癌SMARCA4或SMARCB1/INI1(图8C)缺失表达属于该类肿瘤实体。NUT癌NUT阳性(图8D)同时表达CKpan、p40、p63、CK7及CK20,部分EMA、癌胚抗原(CEA)阳性。这几类肿瘤神经内分泌、黑色素横纹肌标志物阴性。(4)遗传学:未分化癌无特征性遗传学改变,INI1和SMARCA4缺失属于该类肿瘤实体亚型,应区别对待,注意INI1缺失还见于上皮样肉瘤、横纹肌样瘤(中枢、肾等)、50%上皮样恶性外周神经鞘膜瘤、50%的肌上皮肿瘤、20%的骨外黏液软骨样肉瘤、肾集合管癌等;SMARCA4缺失还见于卵巢高钙血小细胞癌、未分化癌、胸部未分化肉瘤、横纹肌样瘤(中枢、肾等)等。NUT癌有NUT-BRD融合基因,NUT的伙伴基因有BRD4和BRD3等。(5)NUT癌细胞幼稚,细胞器少,可见鳞状分化,无桥粒、黑色素颗粒及神经内分泌颗粒。(6)治疗及预后:手术完全切除、放化疗及免疫治疗等。未分化癌(包括INI1和SMARCA4缺失亚型)恶性度高,预后差,总体生存率25%(21%~29%)[13]。NUT癌高度侵袭性,进展快,预后不良。
年龄18~50岁,鼻好发,与EB病毒感染相关。病理学呈小-中等大小淋巴细胞为主,伴坏死、血管侵犯及浸润性生长;免疫表型瘤细胞呈CD3、细胞毒、EBER阳性,CD56阳性或阴性均可。
40岁以上多见,病理学上以中等偏大细胞浸润为主,染色质细腻,可见核仁,瘤细胞凋亡、坏死可形成"星空现象",免疫组织化学瘤细胞CD20、PAX5阳性,Ki-67多>40%。
40岁以上成人好发,多发或孤立性骨或骨外病变,血免疫球蛋白升高或尿本周蛋白升高。病理学上表现为浆细胞样细胞浸润,染色质粗糙,核偏位或"钟表面样",线索是胞质内、核内及间质"嗜酸性小球"。免疫表型浆细胞标志物CD79a、CD38、CD138、PC等阳性。
SRCT属于形态学上具有重叠性的肿瘤家族,除上述肿瘤外,其他也可表现为小圆细胞肿瘤,如异位垂体腺瘤、畸胎癌肉瘤、差分化滑膜肉瘤、血管球瘤、髓系肉瘤、转移瘤等。常规工作方法是:以临床影像学为先导,以病理组织学为基础,以免疫组织化学、分子病理(SSX、EWSR1、HEY1基因检测)、电镜(神经外胚层、横纹肌及黑色素最有效)为手段,最终做出综合诊断。
所有作者均声明不存在利益冲突

























