
20多年前,研究者们首次发现动脉瘤样骨囊肿中存在USP6基因所在染色体位点17p13的恒定易位,随着研究的不断深入和检测手段不断进步,研究者们在多种疾病中发现USP6基因的恒定重排,揭示了其克隆性增殖的本质,加深了对该家族肿瘤的认识。该家族肿瘤不仅具有相同的USP6基因重排,同时在组织形态学上也存在一定重叠,有学者提出应将其归类为同一家族谱系,即USP6基因介导的肿瘤家族。本文不仅对该家族肿瘤的临床病理特征、分子遗传学特征、诊断及鉴别诊断要点进行了回顾,同时对该类肿瘤的特殊发现及现有的相关分子检测技术手段及应用进行了总结。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
泛素特异性蛋白酶6(ubiquitin-specific protease 6,USP6)基因,是一种位于人染色体17p13的嵌合性基因[1, 2]。据报道,USP6蛋白仅在部分间叶源性肿瘤,如骨母细胞瘤、肌纤维瘤中高表达,而在正常组织及上皮源性肿瘤中几乎不表达[3]。1999年,Panoutsakopoulos等首次发现动脉瘤样骨囊肿(aneurysmal bone cyst,ABC)中存在17p13的恒定易位(USP6基因位于该区段)[1]。随后,相同的遗传学改变相继被报道[4]。Oliveira团队的系列研究首次发现在原发性ABC和结节性筋膜炎(nodular fasciitis,NF)中存在频发的USP6重排,证实了其肿瘤性增生本质。2014年,Oliveira等提出将ABC和NF归类为USP6基因介导的肿瘤家族,得到了国际病理界的广泛认可[5]。近年来,随着分子遗传学研究不断深入,该家族成员不断扩大。目前,除原发性ABC和NF外,该家族成员还包括NF的特殊亚型[血管内筋膜炎(intravascular fasciitis,IVF)、骨化性筋膜炎(fasciitis ossificans,FO)及颅骨筋膜炎(cranial fasciitis,CF)]、腱鞘纤维瘤(fibroma of tendon sheath,FTS)、骨化性肌炎(myositis ossificans,MO)、指趾性纤维骨性假瘤(fibro-osseous pseudotumor of digits,FOPD)和良性浸润性肌纤维母细胞肿瘤[6, 7, 8, 9, 10, 11]。该类肿瘤临床症状、发病年龄、性别倾向及发病部位不一,但均具有(肌)纤维母细胞增生性成分及高频USP6重排,易与多种梭形细胞增生性病变混淆。因此,深入了解该家族肿瘤的临床病理与分子生物学特征、诊断及鉴别诊断要点及其相关分子检测技术手段及应用在该类肿瘤临床诊疗过程中至关重要。
本文根据其发病部位将其主要分为USP6基因介导的骨肿瘤(骨原发性ABC)和USP6基因介导的软组织肿瘤,而在USP6基因介导的软组织肿瘤中,根据其是否具有骨化生成分,分为USP6基因介导的不伴骨化生的软组织肿瘤及USP6基因介导的伴骨化生的软组织肿瘤,前者包括NF及其变异亚型(CF和IVF)和FTS,而后者包括MO、FOPD、软组织ABC(soft tissue ABC,STABC)和FO。
根据是否伴发其他基础病变,ABC分为原发性ABC和继发性ABC。据报道,骨原发性ABC是目前仅有的USP6基因介导的骨肿瘤,而继发性ABC不存在USP6重排,提示二者虽然形态高度相似但在遗传学上属于不同类型的病变。原发性ABC是一类快速生长的扩张性良性骨肿瘤,具有局部侵袭性。其可发生于任何年龄段,但更常见于20岁以下人群,女性多见,最好发于长骨干骺端和椎骨后方[12]。极少数可发生于软组织中(此内容将于USP6基因介导的伴骨化生的软组织肿瘤部分进行详述)[12]。最常见的临床表现为疼痛和肿胀,可继发病理性骨折[12]。典型的X线表现为界限清晰的扩张性、偏心性的溶骨性病变。MRI显示伴有分隔的扩张性病变,可见特征性“液-液平面”[12]。目前病灶内刮除仍是其主要治疗方法,但具有局部复发潜能,且多在手术治疗后的2年内复发[12]。据报道,个别病例可出现转移[13]。骨原发性ABC典型组织学多表现为出血性囊腔伴纤维分隔(图1)。纤维分隔主要由增殖活跃的肌纤维母细胞及散在分布的破骨细胞样多核巨细胞组成,内衬骨母细胞的反应性编织骨,并见炎性细胞、外渗的红细胞和增生的毛细血管(图2)。约5%的病例表现为微小囊腔或无囊腔形成,称为实体型ABC[12]。

2004年,Oliveira等[14]发现约70%的骨原发性ABC具有CDH11或USP6重排,并发现CDH11-USP6是其最常见的融合类型。随后越来越多融合配对基因在骨原发性ABC中被报道,如ZNF9、COL1A1、TRAP150、OMD、RUNX2、PAFAH1B1、CTNNB1、SEC31A、EIF1、FOSL2、STAT3、FGFR1、SPARC、USP9X、ASAP1、FAT1、SAR1A、TNC、LUM、PTBP1、SLC38A2、TPM4、DDX17、GTF2I、KLF3、MEF2A、RBM5和VDR[4,15, 16, 17, 18, 19]。此外,需要特别指出的是,继发性ABC不具有USP6重排,有研究认为其仅是形态学上与骨原发性ABC相似,而在本质上并不相同,其可能是各种非ABC骨肿瘤分化的共同终点[14]。
在病理诊断工作中,骨原发性ABC的鉴别诊断主要包括骨巨细胞瘤、骨母细胞瘤、软骨母细胞瘤、纤维结构不良、巨细胞修复性肉芽肿及血管扩张型骨肉瘤等。
1.NF及其变异亚型:NF是一种自限性的(肌)纤维母细胞肿瘤。目前病因尚不明确,有报道显示10%~15%的患者曾有外伤史[20]。NF还包括几个特殊亚型,分别为FO、CF及IVF[6,9,21]。它可发生于任何年龄段,多见于30~50岁的成年人。无明显性别倾向。NF最常见的发病部位为上肢,尤其是前臂,其次为头颈部、躯干及下肢[21]。发生于乳腺、关节内、神经、肠系膜、真皮、胎盘及外阴等的NF也有少量报道[4,21, 22]。NF通常表现为迅速生长的孤立性结节,病程在数周至数月不等。平均直径2~5 cm[21]。其影像学表现多不具特异性。一般采用局部完整切除,且术后罕见复发。复发多考虑为局部切除不完全所致[21]。此外,至今文献报道中仅有个别病例出现转移[23]。组织学上,多数病例与周围分界较清,部分病例可呈浸润性生长。NF由胖梭形的(肌)纤维母细胞组成,呈组织培养样生长模式(图3),间质多呈疏松黏液状。肿瘤细胞内可见核分裂象(图4),但病理性核分裂象极为罕见。常见微囊样变,间质常见红细胞外渗及少量淋巴细胞浸润,偶见多核巨细胞。NF的特殊亚型CF及IVF,与其在组织学上非常相似[21]。长期以来,NF被认为是一种反应性病变,而研究发现92%的NF存在USP6基因重排,揭示了其克隆性增殖的本质。此外,约65%的病例中检出MYH9-USP6融合,后续多项研究也得出相似结论[24, 25]。同时,新的配对基因也不断被发现,包括PPP6R3、CDH11、RRBP1、CALU、CTNNB1、MIR22HG、SPARC、THBS2、COL6A2、SEC31A、COL1A1、EIF5A、COL1A2、PAFAH1B1、NACA、LDHA、SLFN11、PDLIM7、MYL12A、CALD1和TPM4[4,26, 27, 28, 29, 30, 31]。新近,有学者发现在其亚型CF中也存在USP6重排及与MYH9、SPARC、SERPINH1和COL3A1的融合[9]。本团队近期在1例IVF中检测到CTNNB1-USP6的融合,证实了该肿瘤属于USP6基因介导的肿瘤家族成员[6]。此外,在婴儿NF中,我们发现仅约14%的USP6重排的患者为MYH9-USP6融合,提示在婴儿NF可能在遗传学上与经典型NF存在差别[32],但目前病例数仍较少,需进一步扩大病例量进行研究。NF具有细胞丰富、生长迅速、核分裂活跃及浸润性生长等特点,临床及病理医生容易将其误诊为恶性肿瘤[21]。NF需要与多种梭形细胞增生性病变相鉴别,主要包括纤维组织细胞瘤、侵袭性纤维瘤病、炎性肌纤维母细胞性肿瘤、低度恶性肌纤维母细胞肉瘤和黏液纤维肉瘤等。
2. FTS:是一种相对少见的良性(肌)纤维母细胞肿瘤。FTS的病因不明,少数患者曾有外伤史或局部反复摩擦史[33]。FTS发病高峰为20~50岁的成人,男性患者较多见[33]。大部分位于上肢,手指最为常见。较少发生于大关节周围软组织[33]。FTS起源于肌腱或腱鞘,生长缓慢,病程可数月至数年,多小于3 cm。影像学上,多表现为无特异性的软组织包块。局部切除术为FTS最常用的治疗方法,但可能因其与肌腱腱鞘附着紧密或分叶状结构难以完全切除而出现局部复发,未见转移或恶变[33]。根据组织学形态,可将FTS分为经典型FTS和富于细胞型FTS(cellular fibroma of tendon sheath,CFTS),经典型FTS为细胞较为稀疏,可见特征性狭长的薄壁血管或裂隙样间隙。CFTS梭形细胞较为丰富,多与经典型形态同时存在。目前关于FTS的遗传学研究较少。2016年,Carter等[34]通过免疫荧光原位杂交(FISH)检测发现CFTS中存在USP6重排,而经典型FTS中不存在该遗传学改变,该研究认为CFTS可能是“NF腱鞘滑膜亚型”[34]。在CFTS中,MYH9、COL1A1、ASPN、PKM、RCC1、COL3A1、NR1D1、MIR22HG、CAP1、RAB1A、LINC00152、CTNNB1、TNC、SPARC和EMP1与USP6的融合相继被报道[4,7, 8,35, 36]。需指出的是,2021年,Pižem等[7]对18例 FTS进行二代测序,在17例FTS(包括6例经典型和11例富于细胞型FTS)中均检出USP6基因重排。该研究认为无论是富于细胞型还是经典型FTS均应归属于USP6基因介导的肿瘤家族谱系,而FTS可能是包括中-高细胞密度病变到细胞稀疏的病变的连续性肿瘤家系[7]。但有关该家族肿瘤(尤其是经典型FTS)还需扩大样本量行进一步研究。FTS需要与多种好发于上肢肢端,尤其需与手指的纤维母细胞增生性病变相鉴别,主要包括促结缔组织增生性纤维母细胞瘤、腱鞘巨细胞瘤、掌跖纤维瘤病及纤维组织细胞瘤。
USP6基因介导的伴骨化生的软组织肿瘤是一组以增生活跃的(肌)纤维母细胞伴不同成熟度的骨组织化生的病变,主要包括MO、FOPD、STABC和FO。发病原因不明,部分病人有明确外伤史。该类肿瘤在各年龄组均可发病,20~40岁的青年人相对更多见[8,10,37, 38]。男性略多见。除FOPD特发于肢端外,其余病种的病变部位均以下肢居多,其次为上肢、躯干,偶见头颈部。本组病例临床上多表现为伴明显肿胀、疼痛的局部单发的肿块,病程长短不一。影像学上,大多边界清楚,X线和/或CT显示稍低密度肿块,周边见连续或间断的壳状钙化影。MRI病变通常呈混杂信号,周边骨壳为低信号[8,10,37, 38]。USP6基因介导的具有骨样组织的软组织肿瘤为良性病变,目前最常用且有效的治疗方法为局部切除治疗,术后复发病例罕见,复发或与肿瘤未能切除干净相关[38]。组织学上,多数MO存在典型的区带分布结构(图5),病变局部区域可呈NF样形态(图6)。FOPD骨样组织大多凌乱分布于纤维组织中。STABC组织形态与骨内原发ABC相似,可见大小不一的囊腔,囊腔及骨样组织周围可见破骨样多核巨细胞[8,10,39]。FO中常可见NF样形态,骨样组织可随意分布,也可呈区带分布,文献关于FO的病例报道较少,骨样组织的沉积模式多与FOPD相似[8]。
目前,这类具有骨样组织的软组织肿瘤已陆续被证实属于USP6基因介导的肿瘤家族成员。目前已有研究提示,MO/FOPD、STABC和FO中存在USP6重排/融合[8,10,37, 38, 39, 40],近年来,对该类肿瘤USP6配对基因和它们之间亲缘性探索的研究发现,它们中均存在COL1A1-USP6的高频融合,阳性率达67.7%~100%[8,10,37,41, 42]。随着研究不断深入,也有其他的融合类型被发现,如Zhang等[37]在STABC中发现了新的USP6的融合配对基因ANGPTL2,但该类特殊融合的临床病理意义有待进一步分析。
临床工作中,该亚类肿瘤需与多种肿瘤,特别是与具有骨分化的肉瘤相鉴别。主要包括骨外骨肉瘤、恶性外周神经鞘膜瘤伴异源性骨分化、去分化脂肪肉瘤伴异源性骨分化、增生性筋膜炎/增生性肌炎、损伤后骨膜炎、奇异性骨旁骨软骨瘤样增生(Nora病)、甲下外生性骨疣、软组织巨细胞瘤、骨化性纤维黏液样肿瘤等。
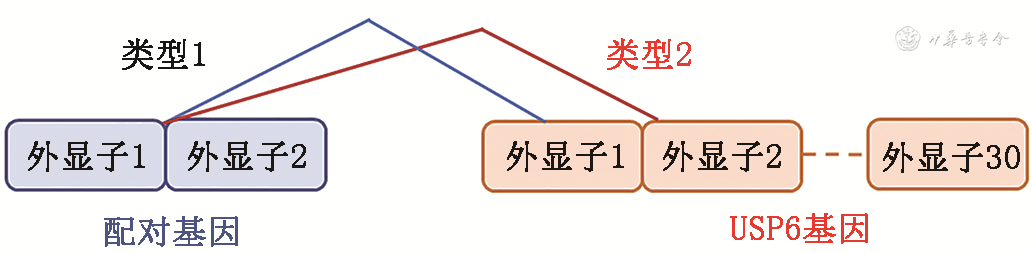
总体来看,在分子水平上,各类肿瘤梭形细胞成分均表现出典型的(肌)纤维母细胞表型,多表达SMA和MSA,一般不表达结蛋白和S-100蛋白。另外,该肿瘤家系存在高频USP6基因重排/融合(图7),且主要融合类型仍以配对基因的非编码序列与保留了的开放阅读框的全基因USP6编码序列(图8)。这种融合可导致USP6的高表达,或为USP6基因介导肿瘤的主要发病机理[21]。但需注意的是,已有研究显示不同的USP6基因介导的肿瘤亚型间主要融合类型存在差异。在ABC中以CDH11-USP6融合为主导,而NF 中MYH9-USP6融合最为常见,COL1A1为USP6基因介导的伴骨化生的软组织肿瘤中最高频的USP6配对基因[8,10,12,37,41, 42]。此外,在FTS中,配对基因MYH9与其他融合配对基因相较相对高频出现,不排除有些MYH9-USP6融合的FTS实际上可能为NF的变异亚型[7, 8,21,35, 36]。另外,需指出的是,长期以来(包括2020年版WHO分类)仍将FO归类于NF的亚型,但FO的配对基因主要为COL1A1而非MYH9,且其主要发病位置为上肢,与NF并不相同,而是与MO相似[8,21]。此外,STABC虽然与ABC在形态学上极其相似,但其常见融合类型为COL1A1-USP6,因而STABC与MO/FOPD等病变可能更具亲缘性,甚至有些学者认为有些MO可能是STABC的早期改变[8,37,39,41, 42]。本组尚未发表的数据也支持这一观点。在临床诊断工作中,各种亚型之间的鉴别虽然具有一定的临床应用价值,但更应注意其与多种恶性肿瘤相鉴别,尤其注意NF与低度恶性肉瘤的鉴别、ABC与血管扩张型骨肉瘤的鉴别、部分伴骨化生的USP6基因介导的软组织肿瘤与具有骨分化的肉瘤鉴别,避免漏诊或误诊带来的严重后果。
随着研究不断深入,近年来,一些具有不典型临床病理和/或分子学特征的病例相继被报道。目前,查阅文献发现13例具有特殊临床病理及分子特征的病例,包括3例骨ABC,6例NF,1例软组织ABC和3例良性浸润性肌纤维母细胞性肿[11,13,15,19,23,26, 27, 28, 29, 30,37]。大部分病例诊断年龄为常见年龄段,少数见于婴幼儿。男性略多于女性。肿瘤大小范围为0.8~27.6 cm,其中5例大于5 cm。影像学上,多数病例表现为浸润性包块(10/11),尤其是2例ABC,一例描述为巨大肿块且影像学可见Codman三角样表现,而另一例出现明显皮质破坏及周围软组织浸润[11,13,15,19,23,26, 27, 28, 29, 30,37]。组织学上,8例未见明显不典型组织学特征;4例NF表现出不同程度的不典型甚至恶性组织学特征,且该4例病灶中均可见病理性核分裂象;1例在活检术后2年内出现轻度不典型核分裂象[11,13,15,19,23,26, 27, 28, 29, 30,37]。另外,所有具有USP6融合基因信息的病例均表现为不常见的配对基因(12/12),包括CALD1、COL6A2、PAFAH1B1、EIF5A、PPP6R3、VDR、RUNX2、COL1A1、COL3A1和ANGPTL2[11,15,19,23,26, 27, 28, 29, 30,37]。值得一提的是,多数病例为USP6不平衡易位(6/10),其中3例伴随USP6基因扩增或拷贝数增多[23,26,30]。目前,有些研究者认为PPP6R3伴基因扩增可能和USP6基因介导肿瘤的恶性行为或恶性转化相关[23,26]。另外,在具有融合位点信息的病例中,仅有1例(本组发表病例)表现出不同寻常的USP6融合位点(exon9,NM_00134284),而大部分病例(11/12)表现为配对基因的非编码区域和USP6 exon1/exon2(NM_004505)的融合[30]。在11例具有随访信息的患者中,1例ABC和1例NF分别出现远处转移[13,23],1例ABC出现复发[15],1例NF和软组织ABC出现了穿刺术后非消退性或侵袭性生长[26,37],但需要指出的是,发生远处转移的患者随访时间长达10年[23]。因此,虽然3例具有随访信息的不典型组织学特征的患者均预后较好(随访时间9~39个月)[27, 28,30],但因其随访时间均较短,不典型性组织学特征的出现是否与恶性生物学行为尚不能定论,应延长随访以获取更多信息。此外,不典型重排或罕见配对基因的出现是否与临床组织学特征相关仍需更多研究。因此,未来的研究中应对这些特殊病例予以更多关注。
FISH及逆转录-聚合酶链反应(RT-PCR)具有快速、简便、成本低等优点,是临床病理工作中最常用的检测基因重排/融合的技术手段,并且在(尤其是早期发表)相关文献中多采用上述方法检测USP6基因的改变。需注意的是RT-PCR只能检测已知融合类型,难以检出未知或隐匿性基因融合。USP6配对基因种类繁多,因此在临床常规工作中多推荐应用USP6易位FISH检测辅助该家族肿瘤的诊断及鉴别诊断[14,23, 24,34]。但是,FISH也具有一定的局限性,例如少数病例信号不典型、个别相邻基因大片段缺失可能引起FISH结果判读的偏倚。目前,随着骨与软组织的基因检测试剂盒的开发及技术的成熟,二代测序技术成本不断降低周期不断缩短,基于二代测序的检测方法在隐匿性重排和新型融合基因的识别中具有很强的应用前景,比如在经典型FTS中,USP6的重排及融合配对基因的发现均受益于二代测序技术的应用[7]。但是无论是传统的FISH和RT-PCR检测技术还是二代测序技术均仅作为USP6基因介导的肿瘤的辅助手段,病例最终诊断仍必需以组织形态学特征为基石。
近年来,随着研究的不断深入,USP6肿瘤家族谱系不断扩大。USP6重排的发现为该家族肿瘤的鉴别诊断提供了有力的依据。越来越多新的配对基因的发现进一步加深了人们对该家族肿瘤的认识。此外,需要特别提出的是,近年来一些具有不典型临床病理和/或分子特征的病例相继被报道,为该家族肿瘤的诊断和治疗带来了新的挑战和问题。我们应进一步加强对这些特殊病例的关注,必要时结合传统检测技术(FISH和RT-PCR)和二代测序手段探索其遗传学特征,进一步分析其临床及分子病理学特征及临床预后之间的关系,避免诊疗不当带来的不良后果。
所有作者声明无利益冲突

























