
伴有破骨样巨细胞的胰腺未分化癌(UCPOGC)是胰腺导管腺癌的一种罕见的特殊亚型。肿瘤成分复杂,其中可见3种细胞类型:肿瘤细胞、单核组织细胞和破骨样巨细胞。肿瘤细胞异型性显著,通常不表达或仅灶性表达上皮标志物,而弥漫表达间叶源性标志物波形蛋白,因此常被误诊为肉瘤;后两者表达组织细胞标志物,缺乏上皮分化。现报道2例UCPOGC,分析临床病理学特征,并查阅相关文献,重点讨论其组织起源及预后相关因素,以提高临床与病理医师对该肿瘤的认识水平。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
例1男,56岁。因皮肤巩膜黄染、腰腹部饱胀不适1个月于2021年2月18日入湖北肿瘤医院。磁共振成像(MRI)示胰头部不均匀稍长T1稍长T2信号肿块影,内多发小囊状长T2信号影,增强实性成分持续中度强化,大小约3.1 cm×2.7 cm,边缘稍模糊(图1)。例2女,53岁。会诊病例,因左腰腹部疼痛1周于2019年11月1日入宜都市中医医院。CT显示左上腹胰胃间隙肿块影。例1行胰十二指肠切除术,例2行胰体尾及脾脏切除术。
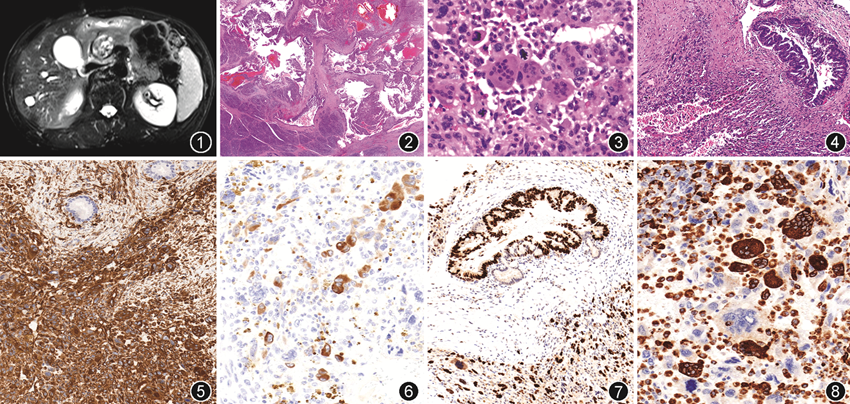
病理检查:(1)大体检查:例1,肿瘤位于胰腺头部,大小3.5 cm×3.5 cm×3.0 cm,切面灰红灰黄,中央可见小囊腔,肿块边界不清,质软。例2,肿瘤位于胰腺体尾部,大小4.5 cm×4.0 cm×3.0 cm,切面灰白灰褐实性,与周围组织界限清楚,质稍硬。(2)镜下观察:低倍镜下,例1肿瘤呈囊实性,例2呈实性,2例肿瘤周围均有较厚的纤维被膜,肿瘤细胞侵犯被膜及被膜外胰腺组织,肿瘤内部可见广泛出血、血湖形成及含铁血黄素沉积(图2),部分区域可见坏死、间质黏液变。高倍镜下,肿瘤细胞多形性及异型性显著,可呈圆形、梭形、多角形,细胞之间黏附松散、界限不清,细胞质中等嗜酸性;细胞核大小不一、形态多样,可呈圆形、卵圆形及多核瘤巨细胞;核染色质粗,核仁明显,病理性核分裂象易见。肿瘤背景中散在分布无异型性的单核组织细胞和破骨样巨细胞(图3),后者细胞核5~30个,大小一致,核染色质细腻,可见小核仁。例1肿瘤内可见脉管癌栓,2例肿瘤中均可见胰腺导管上皮内瘤变(PanIN,图4),邻近肿瘤胰腺组织中均可观察到胰岛(包括紧凑型胰岛和弥漫型胰岛)密度增加,甚至多灶性聚集。2例肿瘤均未侵犯邻近器官,例1送检淋巴结(9枚)未见癌转移,例2未查见淋巴结。(3)免疫表型:2例中肿瘤细胞示波形蛋白弥漫强阳性(图5),广谱细胞角蛋白(CKpan)、细胞角蛋白(CK)19、CK8/18散在弱阳性(图6),CK7、CD68阴性,p53阳性突变型(图7);单核组织细胞和破骨样巨细胞显示CD68(图8)和波形蛋白阳性,其余上皮标记均阴性。PanIN成分CKpan强阳性,波形蛋白阴性,p53亦为阳性突变型。(4)荧光PCR检测结果:例1检测出KRAS基因第2号外显子c.34G>C p.(Gly12Arg)及c.34G>T p.(Gly12Cys)突变,例2检测出KRAS基因第2号外显子c.35G>T p.(Gly12Val)突变;2例均未检测出NRAS基因和BRAF基因V600E突变。
病理诊断:2例均诊断为胰腺伴有破骨样巨细胞的未分化癌。
随访:例1,术后1个月复查腹部CT,未见肿瘤复发或转移,随后行吉西他滨加卡培他滨化疗6个周期,术后随访14个月,无复发或转移征象。例2,术后医生建议出院2周后行化疗治疗,但是患者拒绝,术后随访13个月,无复发或转移。
伴有破骨样巨细胞的胰腺未分化癌(undifferentiated carcinoma of the pancreas with osteoclast-like giant cells,UCPOGC)是胰腺导管腺癌的一种特殊亚型,十分罕见,在胰腺非内分泌性肿瘤中占比不到1%[1]。搜索我院病理系统发现2016年1月至2021年4月诊断的胰腺恶性肿瘤共276例,2例UCPOGC,占比0.7%。UCPOGC常见于中老年人,发病年龄范围广(32~82岁),男性稍多;好发于胰腺体尾部,生长迅速,80%在诊断时最大径超过5 cm,易发生出血、坏死。临床表现多种多样,症状无特异性,常表现为腹痛、黄疸、乏力、体重减轻[2]。影像学检查对于诊断具有提示作用,CT显示边界清楚的囊实性肿块,肿块内部可见分隔、出血、囊性变,增强后实性部分、分隔及边缘强化。MRI表现为混杂信号,T1WI呈低信号影,常伴片状或灶状高信号,提示肿瘤内出血[3]。本组例1镜下见肿瘤内广泛出血及血湖形成,与影像学相符。
UCOGCP镜下特点为异型性不明显的破骨样巨细胞散在分布于显著多形性、呈肉瘤样生长的癌细胞中。破骨样巨细胞表达组织细胞标志物;肉瘤样的癌细胞通常不表达或仅灶性表达上皮标志物,而弥漫表达间叶源性标志物波形蛋白。UCPOGC可伴随其他分化较好的上皮性肿瘤,如胰腺导管腺癌(最多见)、导管内乳头状黏液性肿瘤、黏液性囊腺癌、腺鳞癌等[4]。此外,我们还发现2例肿瘤周围胰腺组织中胰岛聚集,这种现象在以往的报道中未见描述。原因可能是UCPOGC导致胰腺外分泌部萎缩,从而使胰岛数目相对增多。
因UCPOGC特殊的镜下形态,人们一直对其组织学起源充满好奇,早期有研究者通过电镜发现肿瘤细胞缺乏上皮细胞特征,并且免疫组织化学不表达上皮标志物,因此认为此肿瘤来源于间叶组织[5]。近年来,分子检测发现UCPOGC的遗传改变与胰腺导管腺癌十分相似,包括原癌基因KRAS和抑癌基因CDKN2A、TP53、SMAD4突变,这些结果进一步支持了UCPOGC为胰腺导管上皮起源,是导管腺癌的特殊亚型[6]。本组2例也均检测出KRAS基因突变,从分子水平证明了我们的诊断。Sakai等[7]应用显微切割技术发现肿瘤中的破骨样巨细胞无KRAS基因突变,且免疫组织化学CD68阳性,因此认为这些巨细胞是非肿瘤性的、反应性的成分。UCPOGC的发生可能起始于KRAS、CDKN2A、TP53、SMAD4突变等一系列分子事件,这些因素导致PanIN,进一步发展成胰腺导管腺癌,癌细胞发生上皮间质转化(EMT)形成肉瘤样、多形性表型,即为未分化癌,癌细胞产生粒细胞集落刺激因子招募非肿瘤性的破骨样巨细胞,从而形成UCPOGC[4]。本组2例均观察到PanIN,且p53表达模式与UCPOGC肿瘤细胞相同,从组织学上证明了UCPOGC可能由其进展而来。
在鉴别诊断上,当取材有限,切片中没有找到分化较好的导管腺癌成分时,UCPOGC很难与胰腺原发的肉瘤相鉴别,如本组例2原单位诊断即为软组织肿瘤。因胰腺原发的肉瘤极其罕见,所以诊断之前必须完全排除未分化癌,鉴别点包括肉瘤不表达上皮标志物,且无KRAS基因突变。而当UCPOGC合并导管腺癌时需与癌肉瘤鉴别,鉴别点在于后者缺乏破骨样巨细胞。
临床上,胰腺未分化癌是高度恶性的肿瘤,易发生淋巴结转移及血管侵犯。罗秀等[8]发现,UCPOGC与不伴有破骨样巨细胞的未分化癌相比,两者年龄和性别无明显差异,但是前者预后更好,较少发生淋巴管、血管、神经侵犯及淋巴结转移。破骨样巨细胞的出现可能提示机体对肿瘤的免疫反应较强烈,从而UCPOGC预后较普通的未分化癌好。最初人们认为UCPOGC的预后比胰腺导管腺癌差,因为其生长迅速,发现时已是晚期,无法手术切除,即使完整切除后也容易早期复发。然而,新近研究发现UCPOGC预后显著优于导管腺癌,5年生存率>50%[4]。而不伴有胰腺导管腺癌成分的UCPOGC生物学行为更加惰性[9],Luchini等[6]证明单纯性UCPOGC的中位总生存时间为36个月,伴有胰腺导管腺癌成分的总生存期仅为15个月。综上所述,单纯性UCPOGC预后好于伴有胰腺导管腺癌的UCPOGC、导管腺癌及未分化癌,因此,广泛取材对其诊断及判断预后十分重要。如果发现胰腺导管腺癌成分需要在病理报告中列出,以提示临床预后不佳。本组中例1肿瘤全部取材、例2肿瘤广泛取材,均未发现导管腺癌成分,提示预后较好,术后随访1年以上均无复发或转移。
因UCPOGC病例数少,目前对其治疗方式并无统一标准,主要治疗手段是完整切除病灶。化疗和/或放疗的疗效仍有待评估。有学者认为由于UCPOGC是胰腺导管腺癌的一种亚型,因此可以使用标准的化疗方案[4]。PD-1或PD-L1免疫疗法已在多种肿瘤类型中取得成功,已经证明PD-L1在60%~80%的UCPOGC病例的肿瘤细胞中表达,特别是在伴有胰腺导管腺癌的病例中[4]。新近研究表明,免疫检查点抑制剂对胰腺癌原发灶的治疗作用有限,但可能对UCPOGC的肺转移灶有较明显的作用[10]。
所有作者声明无利益冲突

























