
遗传性肿瘤易感综合征(inherited cancer susceptibility syndromes,ICSS)是由于患者胚系基因致病性突变导致其一生中具有多种肿瘤易感倾向的一类疾病。随着二代测序技术在肿瘤基因检测中的应用,越来越多的ICSS为人们所认识。由于这类肿瘤具有与相应散发性肿瘤不同的发病机制、临床表现、病理形态、治疗方式、预后和家系管理措施,因而有必要将其识别出来以促进这类肿瘤的精准诊断、精准治疗和精准预防。但在临床工作中,病理医师由于对患者和家系信息了解不足、对ICSS识别和诊断的必要性认识不够等原因,导致部分遗传性肿瘤被当作散发病例进行诊治,家系高危成员也不能得到有效的预防性监测和管理。鉴于此,本文总结了常见ICSS相关肿瘤的遗传学、临床和病理学特征,并对病理医师如何更好地认识ICSS相关肿瘤提出了几点思考,以期提高病理医师对这类肿瘤的警惕性和识别能力,提高ICSS的诊断水平。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2023年从第1期至第10期共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。
遗传性肿瘤易感综合征(inherited cancer susceptibility syndromes,ICSS)是由于患者胚系基因致病性突变导致其一生中具有多种肿瘤易感倾向的一类疾病。这类肿瘤约占所有肿瘤的10%[1]。随着二代测序等高通量基因检测技术的临床应用,越来越多的遗传易感性肿瘤逐渐被认识和诊断,估计ICSS相关肿瘤的总发病率约为17.5%[2]。而在儿童和青少年人群中,ICSS相关结直肠癌的比例高达35%[3]。我国目前ICSS相关肿瘤的发病率尚不清楚,但我国人口基数大,ICSS并不罕见。
ICSS具有与相应散发性肿瘤不同的发病机制、临床表型、诊疗策略及家系管理措施。而在临床诊疗工作中,临床医师和病理医师对ICSS识别和诊断的必要性认识不够,常忽视ICSS相关肿瘤的临床病理学特征而造成漏诊,导致这部分肿瘤被当作散发性病例进行诊治,患者可能就失去了某些有显著获益的治疗机会,家系高危成员也不能得到有效的预防性监测和管理。因此,在临床诊断工作中如何将这部分肿瘤识别出来并为临床决策提供病理诊断依据,是需要病理医师深入思考和共同探讨的问题。本文拟通过总结文献对ICSS相关肿瘤的遗传学特征、临床和病理学特征的研究进展,以期有助于这类肿瘤的临床识别和精准病理诊断。
1.肿瘤易感基因(cancer predisposition gene,CPG)的分类和致病机制。ICSS多为单基因遗传病,绝大多数为常染色体显性遗传(表1)。发生胚系变异的CPG来自父母一方或双方。常见的CPG分为抑癌基因、代谢基因、原癌基因等。抑癌基因包括:(1)看门人基因(gatekeeper gene):通过调节细胞周期、分裂、分化和凋亡来控制细胞增殖,包括TSC1/2、NF1/2、PTEN、APC、TP53等基因。这类基因功能的失活会导致细胞转化和无限制生长,最终成为肿瘤细胞。(2)看护者基因(caretaker gene):通过识别基因复制过程中出现的错误并进行修复,如错配修复基因(MMR;MLH1、MSH2/6、PMS2)、BRCA1/2、ATM等基因。这类基因功能的失活会引起基因复制错误的累积,导致基因组不稳定性增加和肿瘤的发生。ICSS相关代谢基因包括VHL、延胡索酸水合酶(fumarate hydratase,FH)、琥珀酸脱氢酶(succinate dehydrogenase,SDH;包含A、B、C、D 4个亚基)等,主要参与氧代谢调节,这类基因功能失活后在DNA和蛋白质的表观遗传学修饰、氧化应激、低氧应答等多个维度诱导肿瘤的发生发展。抑癌基因和代谢基因的突变类型包括点突变、缺失突变、插入(移码)突变,产生截短的或无功能的蛋白质产物。常见受累的原癌基因包括RET、MET等。这类基因编码生长因子、生长因子受体等,突变后会导致基因功能增强而促进细胞持续增殖和迁移。近年来,功能相关基因复合体缺陷导致ICSS的发现改变了以往认为ICSS是由单一基因缺陷致病的认识。如Lynch综合征中的错配修复基因复合体(MLH1、MSH2/6、PMS2)、SDH(A、B、C和D 4个亚基基因)缺陷相关ICSS和SWI/SNF(SMARCB1、SMARCA4、SMARCE1、ARID2等亚基基因)缺陷综合征,它们都是由几个功能同源或相关的复合体中某一基因变异引起的[4]。这为将来ICSS新致病基因的寻找和新ICSS的发现提供了思路。ICSS相关肿瘤的发生符合“二次打击”学说,即在生殖细胞水平已经发生的第一次突变使两个等位基因中的一个失去活性,在个体出生后体细胞的另一等位基因发生第二次突变失活即可诱导肿瘤的发生。因此,ICSS相关肿瘤发病较早且易于多发或累及成对的双侧器官。
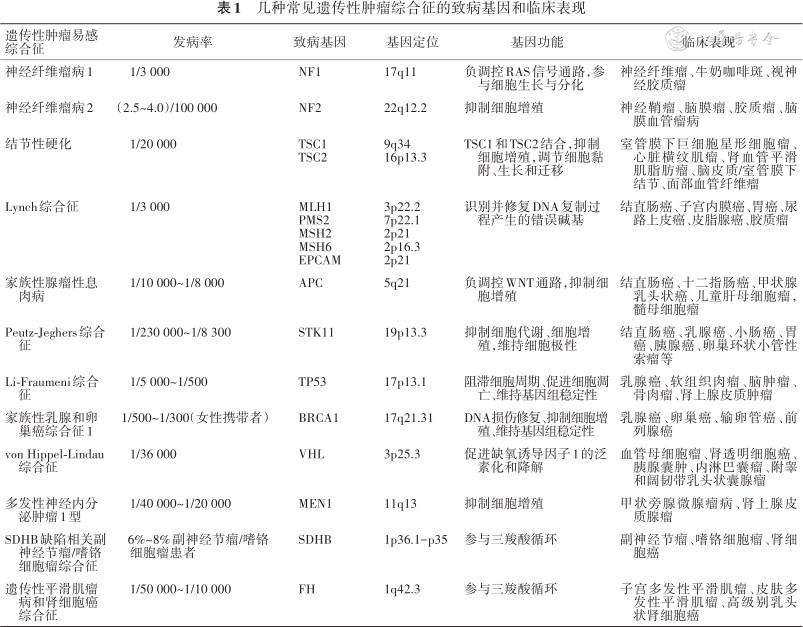
几种常见遗传性肿瘤综合征的致病基因和临床表现
几种常见遗传性肿瘤综合征的致病基因和临床表现
| 遗传性肿瘤易感综合征 | 发病率 | 致病基因 | 基因定位 | 基因功能 | 临床表现 |
|---|---|---|---|---|---|
| 神经纤维瘤病1 | 1/3 000 | NF1 | 17q11 | 负调控RAS信号通路,参与细胞生长与分化 | 神经纤维瘤、牛奶咖啡斑、视神经胶质瘤 |
| 神经纤维瘤病2 | (2.5~4.0)/100 000 | NF2 | 22q12.2 | 抑制细胞增殖 | 神经鞘瘤、脑膜瘤、胶质瘤、脑膜血管瘤病 |
| 结节性硬化 | 1/20 000 | TSC1 TSC2 | 9q34 16p13.3 | TSC1和TSC2结合,抑制细胞增殖,调节细胞黏附、生长和迁移 | 室管膜下巨细胞星形细胞瘤、心脏横纹肌瘤、肾血管平滑肌脂肪瘤、脑皮质/室管膜下结节、面部血管纤维瘤 |
| Lynch综合征 | 1/3 000 | MLH1 PMS2 MSH2 MSH6 EPCAM | 3p22.2 7p22.1 2p21 2p16.3 2p21 | 识别并修复DNA复制过程产生的错误碱基 | 结直肠癌、子宫内膜癌、胃癌、尿路上皮癌、皮脂腺癌、胶质瘤 |
| 家族性腺瘤性息肉病 | 1/10 000~1/8 000 | APC | 5q21 | 负调控WNT通路,抑制细胞增殖 | 结直肠癌、十二指肠癌、甲状腺乳头状癌、儿童肝母细胞瘤,髓母细胞瘤 |
| Peutz-Jeghers综合征 | 1/230 000~1/8 300 | STK11 | 19p13.3 | 抑制细胞代谢、细胞增殖,维持细胞极性 | 结直肠癌、乳腺癌、小肠癌、胃癌、胰腺癌、卵巢环状小管性索瘤等 |
| Li-Fraumeni综合征 | 1/5 000~1/500 | TP53 | 17p13.1 | 阻滞细胞周期、促进细胞凋亡、维持基因组稳定性 | 乳腺癌、软组织肉瘤、脑肿瘤、骨肉瘤、肾上腺皮质肿瘤 |
| 家族性乳腺和卵巢癌综合征1 | 1/500~1/300(女性携带者) | BRCA1 | 17q21.31 | DNA损伤修复、抑制细胞增殖、维持基因组稳定性 | 乳腺癌、卵巢癌、输卵管癌、前列腺癌 |
| von Hippel-Lindau综合征 | 1/36 000 | VHL | 3p25.3 | 促进缺氧诱导因子1的泛素化和降解 | 血管母细胞瘤、肾透明细胞癌、胰腺囊肿、内淋巴囊瘤、附睾和阔韧带乳头状囊腺瘤 |
| 多发性神经内分泌肿瘤1型 | 1/40 000~1/20 000 | MEN1 | 11q13 | 抑制细胞增殖 | 甲状旁腺微腺瘤病、肾上腺皮质腺瘤 |
| SDHB缺陷相关副神经节瘤/嗜铬细胞瘤综合征 | 6%~8%副神经节瘤/嗜铬细胞瘤患者 | SDHB | 1p36.1-p35 | 参与三羧酸循环 | 副神经节瘤、嗜铬细胞瘤、肾细胞癌 |
| 遗传性平滑肌瘤病和肾细胞癌综合征 | 1/50 000~1/10 000 | FH | 1q42.3 | 参与三羧酸循环 | 子宫多发性平滑肌瘤、皮肤多发性平滑肌瘤、高级别乳头状肾细胞癌 |
2.CPG外显率和表现度的差异。CPG在家系内部和家系之间的外显率(penetrance)和表现度(expressivity)有很大差异。如错配修复基因(MSH2、MLH1)和WT1基因变异在结直肠癌和遗传性肾母细胞瘤的外显率分别为90%和30%[5]。同一家系中的Lynch综合征患者临床表型也各不相同。这其中的影响因素包括致病基因的遗传方式,患者性别、年龄,环境和生活方式等。CPG的遗传方式多为常染色体显性遗传,子女有50%的遗传概率,少数为常染色体隐性遗传(如着色性干皮病、结构性错配修复缺陷综合征),子女有25%的遗传概率。在性别因素中,由BRCA2突变引起的家族遗传性乳腺癌中,男性和女性在70岁前乳腺癌的患病率分别为6%和86%。ICSS患者随年龄的增长而肿瘤逐渐显现,如BRCA1/2家族遗传性乳腺癌、多发性内分泌肿瘤综合征(multiple endocrine neoplasia,MEN)2A型、SDHD/SDHB相关家族性副神经节瘤等。而且CPG的突变位点也与发病年龄相关,如BRCA1基因第2号外显子c.185 delAG突变的乳腺癌患病中位年龄为55岁,第11号外显子c.4184 delTCAA突变者为47岁,而第13号外显子重复者则为41岁[6]。此外,环境和生活方式因素,如种族背景、辐射和阳光照射、体力活动、药物和酒精摄入等均会对ICSS的临床外显率和表现度产生显著影响。如在石棉暴露的环境中,BAP1基因胚系突变人群较之未发生突变的人群更早发生恶性间皮瘤,且更容易患腹膜恶性间皮瘤[7]。CPG外显率和表现度的差异不仅解释了为什么ICSS患者父母未受累及,也解释了为什么部分患者携带了CPG却肿瘤不表现为多发或终身不患肿瘤。
1.肿瘤发病年龄早。发病年龄早是大多数ICSS的主要特征之一。遗传性弥漫型胃癌平均发病年龄38岁,而我国散发性胃癌多发生在50岁以后[8]。BRCA1/2突变的家族遗传性乳腺癌患者平均发病年龄较散发性乳腺癌患者早5~8年。而散发性乳腺癌患者仅17%左右为早发性乳腺癌[9]。Von Hippel-Lindau(VHL)综合征患者肾细胞癌平均发病年龄为40~45岁,比散发性肾癌平均发病年龄约早20年。Lynch综合征患者中,结直肠癌平均发病年龄为40~50岁,而散发性结直肠癌患者多>60岁;95%的子宫内膜癌患者发病年龄<65岁,60%的患者<50岁,平均年龄46~49岁,较之散发性Ⅰ型和Ⅱ型子宫内膜癌分别年轻6~10岁及15~20岁[10];卵巢癌平均年龄为42~49岁,散发性卵巢癌中位发病年龄为60岁。
2.肿瘤的家族聚集性。肿瘤的家族聚集性是ICSS的另一主要特征。如果一个家庭中≥2位一级和/或二级亲属患有肿瘤,其中至少1例患者发病年龄<50岁,或患有特定的肿瘤组合,均应怀疑ICSS,需要进一步行基因筛查和遗传咨询。在临床工作中应重视患者家族史的问诊。通过一级亲属、二级亲属、三级亲属的顺序逐步询问,了解这些亲属的健康状况、是否曾患肿瘤及患病年龄和治疗信息、是否健在及死亡年龄和原因等,以求更全面、完整地获得患者家族史信息,减少漏诊。
3.具有特定的好发部位。部分ICSS相关肿瘤较之散发性肿瘤具有特定的发生部位。Lynch综合征相关结直肠癌中约60%发生在右半结肠,而相关子宫内膜癌则好累及子宫下段。结节性硬化症相关室管膜下巨细胞星形细胞瘤主要位于侧脑室侧壁邻近Monro孔。Cowden病患者常发生小脑发育不良性节细胞瘤。VHL综合征患者血管母细胞瘤常见于小脑、脊髓和视网膜部位。
4.累及成对的双侧器官。成对器官发生同时或异时性肿瘤应怀疑ICSS。国内研究数据表明,有乳腺癌家族史的BRCA1/2突变乳腺癌患者对侧乳腺癌发病风险约27%[11]。家族性甲状腺非髓样癌患者双侧甲状腺患癌比例达44%[12]。30%~50%的双侧肾上腺嗜铬细胞瘤存在致病性胚系突变,可见于MEN2A/2B型、VHL综合征、NF1综合征、SDH缺陷综合征等[13]。其中,双侧附睾囊肿和囊腺瘤、双侧耳内淋巴囊瘤均提示VHL综合征。
5.同一类型肿瘤的多灶性原发。同一类型肿瘤累及身体多个部位时应怀疑ICSS。NF1可表现为身体皮肤表面数个到上百、上千甚至全身上下长满大小不一、难以计数的神经纤维瘤。NF2相关神经鞘瘤常发生于皮肤,也可同时或异时性发生于多处颅神经和脊神经。Lynch综合征相关结直肠癌切除后约32.2%患者会再发生一次以上的异时性结直肠癌[14]。在SDHD突变相关的遗传性副神经节瘤/嗜铬细胞瘤综合征中,60%的患者表现为头颈部多发性副神经节瘤。在临床诊疗实践中应认识到这些多灶性肿瘤与遗传的潜在关联。如果将这些多原发的肿瘤误认为是转移性肿瘤可能会导致肿瘤的过度分期和过度治疗。
6.同时或异时性多种肿瘤或非肿瘤病变的组合。ICSS常同时或异时性发生多种肿瘤或非肿瘤性病变(表1)。Lynch综合征患者以结直肠癌、子宫内膜癌最为多见,此外还可发生胃癌、尿路上皮癌、卵巢癌、神经胶质瘤、皮肤肿瘤等多系统的肿瘤。VHL综合征临床表现为中枢神经系统血管母细胞瘤、肾细胞癌、胰腺囊肿、肾上腺嗜铬细胞瘤、内耳内淋巴囊肿瘤和附睾囊腺瘤。BRCA1/2突变的家族遗传性乳腺癌综合征患者主要表现为乳腺癌和卵巢癌。NF2表现为双侧听神经瘤、外周神经鞘瘤和脑膜瘤。MEN 1型表现为全身多灶性内分泌肿瘤,可累及甲状旁腺、内分泌胰腺、垂体前叶、肾上腺皮质等器官。因此,患者多种肿瘤或非肿瘤病变的组合都提示ICSS,病理报告对此应予以注释和提示。但在临床工作中,许多并发肿瘤可能无法找到它们之间的联系,有些可能是常见恶性肿瘤的巧合,另一些可能与环境致病因素或尚未确定的遗传因素有关。
7.伴随身体异常。部分ICSS伴有先天性骨骼、皮肤等身体异常表现,可作为识别和诊断ICSS的线索。部分CDH1基因突变相关的遗传性弥漫型胃癌家系成员同时合并唇裂或腭裂的先天性畸形[15]。Birt-Hogg-Dub(BHD)综合征患者除发生肾细胞癌外,90%的患者可发生皮肤纤维毛囊瘤。结节性硬化症患者可表现为鼻面部多发血管纤维瘤(图1)、指甲纤维瘤、皮肤鲨鱼皮斑等皮肤异常。黑斑息肉综合征(Peutz-Jeghers综合征)常见面部、口唇周围、手掌(图2)、足底等皮肤、黏膜色素沉着。此外,皮肤牛奶咖啡斑可见于10%的正常人,但多量的牛奶咖啡斑应怀疑NF1、Turcot综合征、共济失调性毛细血管扩张综合征、结节性硬化症等。
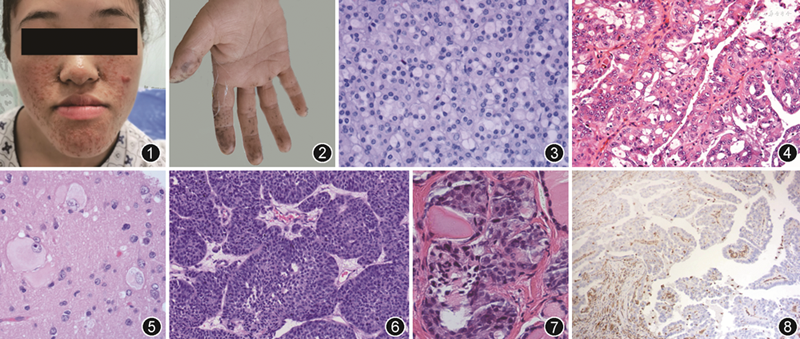
1.提示性的形态学特征。部分ICSS相关肿瘤具有与相应散发性肿瘤不同的病理学形态表现。SDHB缺陷胃肠道间质瘤细胞多表现为上皮样细胞形态,呈丛状或多结节样生长方式,常见淋巴管内瘤栓;SDHB缺陷肾细胞癌胞质丰富,呈空泡状、絮状或羽毛状(图3)。遗传性平滑肌瘤病和肾细胞癌(hereditary leiomyomatosis and renal cell carcinoma)综合征相关平滑肌瘤镜下可见鹿角状血管,胞质可见嗜酸性包涵体,可伴有奇异核和/或不典型性,核内可见嗜酸性核仁伴周围空晕;相关肾细胞癌可呈管状乳头状形态,瘤细胞胞质丰富、细胞核大、伴包涵体样嗜酸性核仁(图4)。结节性硬化症相关癫痫脑组织内可见气球样巨细胞(图5),胞质嗜酸性,核常偏位,呈双核或多核,可有明显核仁。Peutz-Jeghers综合征消化道息肉中心平滑肌肌束呈树枝状延伸至息肉顶部,每个分支表面均有黏膜被覆;卵巢可以发生环状小管性索肿瘤,肿瘤细胞巢由单层和复层环状小管组成,中央为玻璃样小体,细胞巢周边绕以基底膜样物质。BRCA1突变的家族遗传性乳腺癌镜下表现为肿瘤“推挤性”边缘、淋巴细胞浸润,细胞核级别高、核分裂象易见,约15%病例呈“髓样癌”形态[16]。Lynch综合征相关结直肠癌肿瘤边缘呈膨胀性浸润,伴显著的上皮内淋巴细胞浸润;相关尿路上皮癌肿瘤呈内翻性生长方式(图6),边缘呈膨胀推挤性,瘤细胞高核级但缺乏多形性。此外,丛状神经纤维瘤、软组织巨神经纤维瘤几乎仅见于NF1患者。镜下见迂曲、膨大的神经纤维束呈丛状或多结节状排列,间质伴黏液样变性。需要注意的是,由于散发性肿瘤的高占比,加之某些致病性基因的表达缺陷属于体细胞异常而非胚系异常(如结直肠癌MLH1基因启动子区高甲基化)等原因,上述形态学线索对ICSS的诊断具有提示作用,但并非特异性的。
2.提示性的肿瘤前体病变。CDH1基因胚系突变的遗传性弥漫型胃癌镜下表现为胃黏膜腺体原位病变伴周边正常黏膜内Paget样分布的印戒细胞癌[17]。家族性胃肠道间质瘤可伴随胃肠壁Cajal间质细胞增生[18]。而甲状腺滤泡旁细胞(C细胞)增生(图7)、肾上腺髓质增生提示MEN2[19]。对这些前体病变和微小早期肿瘤的识别是诊断ICSS的有用线索。
3.提示性的免疫组织化学指标。某些基因胚系变异可导致肿瘤中该基因表达缺失,应用免疫组织化学方法通过评估这类蛋白质的表达状态对ICSS的诊断具有提示作用。由SDH亚基基因突变引起的家族性副神经节瘤、嗜铬细胞瘤、肾细胞癌、胃肠道间质瘤等肿瘤表现为SDH(A、B、C和D)4个亚基之一表达阴性。横纹肌样瘤易感综合征相关的脑非典型畸胎瘤样/横纹肌样瘤表现为SWI/SNF复合物中SMARCB1(INI1)或SMARCA4(BRG1)表达缺失。Lynch综合征相关结直肠癌、子宫内膜癌等表现为MMR蛋白(MLH1、PMS2、MSH2、MSH6)表达缺失。遗传性平滑肌瘤病和肾细胞癌综合征相关平滑肌瘤和肾细胞癌表现为FH表达缺失(图8)。由于免疫组织化学染色结果受组织固定、预处理、抗体克隆号不同等多种因素影响,对上述表达缺失蛋白质免疫组织化学染色结果的判读及解释需注意以下几点:(1)观察组织内正常上皮、血管内皮细胞、纤维细胞、淋巴细胞等内对照是否为阳性,以避免出现假阴性判读。(2)当基因突变引起该蛋白功能丧失却保留了其抗原性,免疫组织化学可显示假阳性染色结果。(3)某些蛋白质表达阴性可能是由于体细胞突变或启动子区甲基化(如MLH1基因)等表观遗传学机制导致的。后两种情况需进一步行胚系基因检测确认。
1.具备一定的遗传学和遗传咨询知识。个体的遗传特性是ICSS相关肿瘤发生的主要风险因素。病理医师应具备一定的遗传学知识,通过收集患者的肿瘤史、家族史资料,绘制家系图谱,分析遗传因素在肿瘤发生中的作用,探究可能的致病基因、基因变异及其遗传方式。这不仅有助于ICSS相关肿瘤的临床和病理诊断,还将有助于发现新的ICSS。此外,病理医师需要具备一定的遗传咨询知识。遗传咨询是通过评估家族肿瘤史、个人肿瘤史、环境暴露因素、生活习惯、基因检测结果等信息以确定肿瘤发生风险、遗传方式和基因检测技术,提供相应肿瘤的治疗和预防管理方案,帮助咨询者作出知情、自主的医疗保健决策和生育选择,并提供情感和心理支持以及道德指导[20]。目前,我国肿瘤遗传咨询工作缺乏宣传,咨询者主观能动性不强,遗传咨询服务队伍建设有待加强。病理医师可参加国内遗传咨询培训并进行职业资格认证,并尝试开设遗传咨询门诊,以帮助患者解决肿瘤遗传咨询方面的问题。
2.具备一定的分子生物学技术知识。病理诊断中对可疑的ICSS需进一步通过基因检测来确诊。由于二代测序技术具有高通量的特点,目前越来越广泛地应用于遗传病的临床筛查领域。基于二代测序平台的测序技术包括:靶向基因测序、全外显子组测序(whole exome sequencing)和全基因组测序(whole genome sequencing)。这些测序技术均有相应的适应证、优势及局限性。如人类基因组中外显子占总序列的1%~2%,却包含了85%的疾病相关基因突变。全外显子组测序只需针对外显子区域的基因序列进行测序即可实现更高的测序深度,更加简便、经济、高效。但全外显子组测序对结构变异与非编码区变异的研究具有局限性,相关疾病可通过全基因组测序进行检测。因此,病理医师应深入分析患者临床表型,正确选择合适的基因检测策略以提高检测阳性率并优化检测成本。此外,高通量测序在提高诊断阳性率的同时也产生了大量的基因变异数据。如何结合临床表型对这些数据进行准确解读和临床意义分级是ICSS肿瘤基因诊断的重大挑战。目前我国承担基因检测工作的多为第三方商业性基因检测机构,各实验室的基因检测技术和结果分析水平参差不齐。病理医师需提高对患者表型数据的提取能力、对基因检测报告的分析和甄别能力。未来ICSS相关肿瘤的病理诊断可尝试整合肿瘤的组织学特征和分子遗传学信息,为临床和患者提供更个体化的病理诊断报告。此外,将基因检测结果与肿瘤形态学相结合不仅有助于阐明ICSS相关肿瘤的形态学特征,也有助于发现新的致病性基因、基因新的致病性变异或新的ICSS。
3.了解ICSS相关肿瘤的临床治疗、预防性监测和生育健康管理知识。由于ICSS具有多原发和同时或异时性发生多种肿瘤的特点,部分患者可以采取易受累器官的预防性切除。如Lynch综合征患者结直肠和子宫的预防性切除、BRCA1/2突变患者输卵管和卵巢的预防性切除等。另外,某些ICSS相关肿瘤具有特殊的辅助治疗方法。如Lynch综合征相关结直肠癌不能从氟尿嘧啶单药辅助化疗中获益,对于晚期患者推荐一线使用免疫检查点抑制剂治疗[21]。对于家系成员胚系致病基因携带者,需要了解ICSS相关肿瘤谱系的家系管理措施,有利于肿瘤的早发现和早治疗,提高患者生存率和生存质量。此外,对于有生育需求的胚系突变携带者建议进行产前诊断和辅助生殖,包括胚胎植入前遗传学诊断/筛查并进行优生优育咨询,选择正常或不携带相关突变基因的胚胎移植,从源头上阻断致病性基因遗传给后代[22]。因此,ICSS相关肿瘤临床治疗和家系管理的特殊性为肿瘤的病理诊断提出了更高的要求。
综上所述,由于病理科负责医院所有科室的活检和手术标本诊断,在疾病诊断中居于中心地位,能够对患者同时或异时性多器官的不同肿瘤进行综合分析,提出这些肿瘤与遗传之间潜在的关联性。而且病理医师兼具临床医学和基础医学专业知识,能够对可疑的ICSS相关肿瘤通过免疫组织化学、基因测序等实验方法在蛋白质和DNA水平上进一步验证和诊断。因此,临床病理医师在ICSS相关肿瘤的识别和诊断中具有重要作用。目前国内对ICSS的临床诊疗和家系管理日益重视,已制定了《中国家族遗传性肿瘤临床诊疗专家共识(2021年版)》,涉及家族遗传性胃癌[8]、乳腺癌[9]、结直肠癌[21]等7类癌种。这也对病理医师的肿瘤诊断工作提出了更高的要求。WHO人类肿瘤分类系列丛书中均有对各系统ICSS的详细描述。国内病理医师需要了解各种ICSS的致病基因、遗传特征、临床表型和肿瘤形态。在病理诊断中应注意患者的发病年龄、过去史、家族史、肿瘤形态等临床和病理学线索,综合运用免疫组织化学、基因检测等辅助手段,对可疑的ICSS应在病理报告中进行怀疑和注释,这将有助于患者肿瘤的精准诊断、精准治疗以及家系成员肿瘤的精准预防。
姚志刚, 覃业军. 常见遗传性肿瘤易感综合征的临床和病理诊断现状及存在问题的思考[J]. 中华病理学杂志, 2023, 52(5): 538-543. DOI: 10.3760/cma.j.cn112151-20220829-00744.
河北医科大学第二医院病理科张祥宏教授对本文提出了许多宝贵意见和建议
所有作者声明无利益冲突
1. 下列哪项不都是抑癌基因()
A. TSC1,NF2,PTEN,APC
B. Rb,P53,APC,MSH2
C. LKB1,BRCA1,PTEN,MET
D. TP53,CDH1,MEN1,SMAD4
2. 下列哪项遗传性肿瘤易感综合征患者后代的遗传概率为25%()
A. Lynch综合征
B. 结构性错配修复缺陷综合征
C. 神经纤维瘤病1
D. 结节性硬化
3. 下列哪些属于遗传性肿瘤易感综合征的临床特征()
A. 肿瘤发病年龄早
B. 肿瘤具有家族聚集性
C. 易患同时或异时性多种肿瘤或非肿瘤病变
D. 以上都是
4. 下列遗传性肿瘤易感综合征对应的基因异常不正确的是()
A. Peutz-Jeghers综合征:APC
B. 遗传性平滑肌瘤病和肾细胞癌:FH
C. 结节性硬化症:TSC1
D. Birt-Hogg-Dub(BHD)综合征:FLCN
5. 下列哪项分子生物学技术不属于遗传性肿瘤易感综合征的检测技术()
A. 一代测序
B. 二代测序
C. Western blot
D. 多重连接探针扩增技术(MLPA)

























