
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
先天性肌无力综合征(congenital myasthenic syndrome,CMS)是一组因基因缺陷所致的神经肌肉接头功能障碍性疾病,常见于新生儿或婴儿。1949年Levin首次应用本病名[1]来描述一对母亲健康而于婴儿早期起病的兄妹,并以此同新生儿重症肌无力相鉴别。国内少见该病相关报道。我们报道1例经基因证实的CMS病例。
患者男性,16岁,因"四肢无力"于2008年4月2日就诊于我院。患者自幼较高强度运动后易疲劳。2007年冬(16岁)出现四肢上抬费力。2008年2月起出现轻度活动后疲劳感明显,小跑后易出现全身无力,难以起立,休息后部分好转,无晨轻暮重。外院诊断为"重症肌无力可能性大",给予溴吡斯的明治疗,无力症状加重,停用后好转。既往史无特殊。否认类似家族史。2008年4月入住我院神经科。入院体检:患者身材矮小,颈部力弱。四肢肌力近端Ⅲ+~Ⅵ级,远端Ⅴ-级。四肢腱反射对称减低。余神经系统体检未见明显异常。入院后完善肌酶谱、乙酰胆碱受体抗体、组织免疫相关抗体检查,未见异常。患者新斯的明试验阴性。肌电图示肌源性损害;重复神经电刺激示低频刺激波幅递减;神经传导测定可见重复的复合肌肉动作电位(R-CMAP)波,低频重复刺激后消失;单肌纤维肌电图示右伸指总肌jitter值增宽。肌肉活体组织检查光镜示少数肌纤维轻度萎缩;电镜示弥漫性肌管轻度扩张。入院后曾试验性给予溴吡斯的明60 mg,3次/d,患者出现眼睑下垂及呼吸困难,逐渐停用溴吡斯的明;并给予丙种球蛋白20 g/d ×5 d、泼尼松30 mg/d治疗后,患者无力症状明显好转。结合患者病史、辅助检查,诊断为"CMS可能性大",患者于2008年6月自行出院。
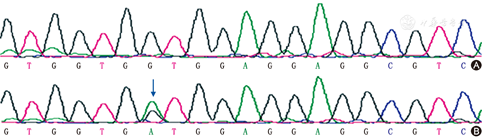
患者出院后未予任何治疗,无力症状轻重程度相差较大,休息及天气暖和时好转,紧张及天气寒冷时加重。2014年3月再次入住我科。体检基本同上次住院。复查肌电图示神经传导测定可见R-CMAP波,低频重复刺激后消失(图1)。送外周血标本行CMS相关基因检测。检测结果示:COLQ基因(NM_080539)外显子中存在1个错义突变:C.175C>T(p.Pro59Ser),为杂合突变(图2),测序深度50/44(0.47)。经文献复习[2],该突变与终板乙酰胆碱酯酶缺乏症相关。同时,对患者父母进行该位点检测,患者父亲存在相同杂合突变,但不存在无力表现。取得患者知情同意后给予特布他林2.5 mg 2次/d治疗,患者自觉无力好转并出院。出院后继续服用特布他林3个月,活动耐量较前好转,后自行停用。目前随诊中。

CMS为罕见的神经肌肉接头疾病,多为常染色体隐性遗传,仅慢通道综合征为常染色体显性遗传。根据其损害部位,可分为突触前缺陷(7%~8%)、突触缺陷(14%~15%)、突触后缺陷(75%~80%)[3,4,5,6,7,8]。因基因突变及分子机制不同,可分为多种亚型[9]。
在发现的突变基因中,CHRNE基因突变最为常见[10],亦为第一个被发现的突变基因。该基因突变可致乙酰胆碱酯酶缺失症[11],是突触型CMS最常见的形式,为常染色体隐性遗传,多为纯合突变或复合杂合突变。我们报道的患者为单位点杂合突变,其遗传模式不符合常染色体显性遗传模式,亦难以用常染色体隐性遗传来解释。COLQ基因突变类型与表型之间没有明确的相关性,额外的修饰因子或者环境因素的存在可能参与其中。既往报道[12,13]曾提及杂合子携带者可出现先天性上睑下垂、肌电图改变等轻度无力的表现,提示COLQ基因携带者可出现肌无力症状。有报道称具有相同基因型的兄弟之间肌无力的程度不同[14],提示相同的基因型可有不同的表型。患者与其父亲基因型相同而肌无力表现不同可用上述说法来解释。
CMS具有一些共同的临床特点:(1)起病早,多于婴儿期或出生时发病;(2)肌肉易疲劳,症状呈波动性;(3)多出现肢体无力、喂养困难、呼吸困难等表现;(4)可有类似肌病的表现如肌肉萎缩;(5)重症肌无力相关抗体阴性[15]。但因分子遗传机制的不同,其发病年龄、症状、受累范围及治疗反应各异,疾病进程及严重程度高度可变。同一患者在病程的不同阶段也可以存在不同表现。一些特殊的临床症状可提示某种疾病或某个基因的缺陷,如慢通道综合征、乙酰胆碱酯酶缺乏应用胆碱酯酶抑制剂效果差;可出现特征性的R-CMAP波。
在治疗方面,患者的治疗反应取决于不同亚型,除慢通道综合征、乙酰胆碱酯酶缺乏症、DOK7缺陷、LAMB2缺陷外,其余一线治疗均可应用溴吡斯的明。同时,越来越多的研究结果表明CMS获益于β受体激动剂[16]。

























