
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肝豆状核变性(hepatolenticular degeneration, HLD)又名Wilson病,为常染色体隐性遗传的铜代谢障碍疾病。Wilson病在世界范围内的发病率为1/3万~1/10万[1]。患者由于铜在不同部位的异常沉积,在临床上表现为进行性加重的以肝硬化、锥体外系症状、肾脏损害和角膜病变为主的多系统损害。由于HLD是至今少数的可治疗的遗传代谢性疾病之一,早期的诊断对临床治疗及预后很有帮助。而MRI对于Wilson病的早期发现具有重要的意义。现就1例肝豆状核变性患者少见MRI表现报道如下。
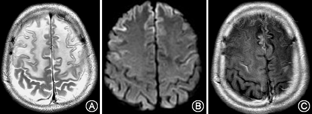
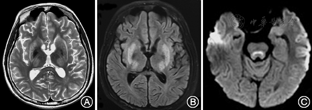
患者男性,16岁,因"阵发性木讷,四肢僵硬、构音障碍10个月"于2016年8月29日入院。患者于10个月前无明显诱因出现阵发性木讷,目光呆滞,持续数十秒后自行好转,而后出现上肢不自主抖动,呈持续性,逐渐出现双上肢先后僵硬,呈屈曲状,不能伸直,后双下肢僵硬,行走不稳。构音障碍,言语不能,饮水呛咳,症状呈进行性加重,自起病来,无发热、头痛,无晕倒、口吐白沫,无肢体偏瘫,无大小便失禁。既往史家族史无特殊。神经系统体检:意识清楚,认知功能尚可,言语不能;K-F环(+);四肢肌张力高,双上肢肌力Ⅳ级,双下肢肌力Ⅴ级,四肢腱反射(+),病理征阴性。生化检查:尿铜792.28 μg /24 h(正常值15~30 μg/24 h);血清铜蓝蛋白测定0.12 g/L (正常值0.2~0.6 g/L);尿常规:隐血3+,蛋白质(±),红细胞832.92个/μl[正常值0~5个/μl];血常规:白细胞计数3.51×109/L[正常值(4~10)×109/L],红细胞计数4.08×1012/L[正常值(4.09~5.74)×1012/L],血红蛋白123 g/L(正常值131~172 g/L),血小板计数78×109/L[正常值(85~303) ×109/L]。凝血四项:纤维蛋白原浓度1.43 g/L(正常值2~4 g/L),凝血酶原时间14.6 s (正常值9~13 s ),国际标准比率1.25 (正常值0.8~1.2),凝血酶原活动度60.1% (正常值70%~150%)。肝功能:碱性磷酸酶210.53 U/L(正常值34~104 U/L),谷氨酰转酞酶58.97 IU/L(正常值5~55 IU/L),纤维结合蛋白127.03 mg/L(正常值180~280 mg/L)。粪便常规+潜血、肾功能、电解质、肌酶谱、C反应蛋白、红细胞沉降率、血脂、餐后2 h血清葡萄糖、叶酸、维生素B12、游离甲状腺素测定等未见异常。患者心电图示窦性心律,大致正常心电图。脑电图:间歇期,左额区见较多中波慢波及尖波,尖慢波;诊断提示:间歇期,痫样放电及慢波,脑区性,左额著。上腹部CT提示肝硬化、脾肿大。颅脑磁共振平扫:双侧额、顶、颞叶脑沟变窄,脑组织肿胀,呈稍长T1稍长T2信号(图1A),DWI呈脑回样高信号(图1B),双侧豆状核、丘脑及外囊、胼胝体、脑干背侧呈片状稍长T1、稍长T2信号,双侧苍白球及小脑齿状核呈等T1、短T2信号(图2)。颅脑增强扫描:双侧额顶叶脑回线样明显强化(图1C)。
在患者及其家属的知情同意下,对患者进行基因检测。在该患者ATP7B基因12号外显子(图3A)检测到错义变异c.2731G>A;p.A911T(杂合),据目前文献和数据库检索,暂不明确是否具有病理意义。在8号外显子(图3B)检测到错义突变c.2333G>T; p.R778L(杂合),为文献报道过的病理性突变位点。

患者青少年男性,癫痫起病,伴有肌张力障碍,角膜K-F环阳性。尿铜高,血清铜蓝蛋白低,镜下血尿,微量蛋白尿,肝硬化等。结合病史、体检及辅助检查结果,根据HLD的诊断标准[2],在我院诊断为HLD明确。经半年随访观察,患者在安徽中医药大学神经病学研究所附属医院(中国HLD中心)进一步驱铜、抗癫痫、护肝等对症治疗后,症状好转。
该患者于省内外就诊,历经10个月未能及时确诊,然而HLD早期识别和及时治疗对患者生活质量的恢复和提高是至关重要的。HLD在MRI表现上有多种表现,且其颅脑磁共振特征性改变与神经系统症状和疾病严重程度相关密切[3]。国外一项纳入100例HLD患者的研究[4]发现,磁共振表现为特征性苍白球T2低信号(34%)、"大熊猫脸"征(12%)、纹状体T1高信号(6%)、脑桥中央髓鞘征(7%)、明亮的屏状核征(4%)。磁共振上表现为信号强度改变和脑实质萎缩都有相关报道[5]。而且疾病的不同发展阶段和磁共振成像系统的特性都可能会影响病变的表现[6],造成了HLD在磁共振影像学表现上的多样性。
本文HLD患者颅脑MRI表现为相对非典型的双侧额、顶、颞叶脑沟变窄,脑组织肿胀,呈稍长T1稍长T2信号,弥散加权成像呈脑回样高信号,增强提示双侧额顶叶脑回线样明显强化以及HLD典型的表现如双侧豆状核、丘脑及外囊、胼胝体、脑干背侧呈片状稍长T1稍长T2信号。脑回线样强化与血脑屏障破坏有关,且脑实质表面强化通常系血管性疾病或炎症性疾病所致[7]。患者有HLD罕见的癫痫发作[8],癫痫所致血管舒张也可导致脑回线样强化。结合国外1例额叶脑回线样强化伴有癫痫发作的HLD患者在抗癫痫治疗1个月后,癫痫症状和脑回线样强化消失,然而额叶皮质下白质影像学改变仍存在且范围相对更大[9],可以推测脑回线样强化与癫痫发作相关,可能是癫痫发作导致脑回线样强化,加剧了脑白质病变。相关研究还发现[8],HLD患者中有癫痫发作者比无发作者有更多的脑白质病变,特别是额叶皮质下病变与癫痫发作呈显著相关。且提示未干预控制的HLD患者癫痫发作,MRI上可能出现脑白质空泡。癫痫发作不是HLD患者常见表现,其往往合并有广泛白质病变[10,11]。目前癫痫与皮质下白质损害因果关系暂不明确。由于HLD影像学存在多样性,诊断应该拓宽思路,还需结合临床病史,及其余辅助检查。
锥体外系症状是脑型和混合型HLD的主要症状,MRI作为锥体外系疾病首选鉴别影像学检查,在其中发挥重要作用。HLD在影像学上的多样性使同样表现为锥体外系症状的相关疾病的鉴别造成一定困难。国外一项研究共回顾性分析了100例早发性锥体外系疾病,发现以下MRI特点能明确区分HLD和其他早发性锥体外系疾病:(1)基底节、丘脑和脑干都有信号改变;(2)"大熊猫脸"征;(3)中脑顶盖的改变;(4)类似于脑桥中央髓鞘溶解症的变化[12]。上述特殊MRI特点给我们对锥体外系症状表现的HLD诊断提供了参考。
众所周知,基因全序列检测是基因诊断中最准确、最直接的的方法,HLD基因(又称ATP7B)全序列检测是目前HLD基因诊断的"金标准"。国内学者吴志英等[13]对多例肝豆状核变性患者进行了HLD基因全长外显子的突变分析,证实了8号外显子的Arg778Leu和12号外显子Thr935Met为我国HLD基因的2个突变热点。且据有关研究表明,我国HLD基因突变以第8、12、13和16号外显子为主,占70%以上[14,15]。我们用PCR和基因测序的方法对患者ATP7B基因编码区外显子8、12、13和16进行了突变分析,涵盖了该范围内的点突变、插入和缺失型突变。发现该患者检测到的1个致病性杂合突变位c.2333G>T;p.R778L,同时检测到1个杂合的未知病理意义的变异位点c.2731G>A;p.A911T,目前尚未见文献报道。
HLD为常染色体隐性遗传病,患者父母双方未患病,无家族史。结合相关研究[13]认为,我国HLD基因突变是以少数几个热点突变和广泛罕见突变为特征,我们应该可以推测,该HLD患者的未知病理意义的变异位点c.2731G>A;p.A911T(杂合)可能为罕见致病性基因突变,与c.2333G>T; p.R778L(杂合)组成复合杂合突变导致患者患病。而且我们通过基因预测软件Polyphen2(http://genetics.bwh.harvard.edu/pph2/)和SIFT(http://sift.jcvi.org/)对未知病理意义的c.2731G>A;p.A911T错义突变进行功能预测,发现Polyphen2预测结果提示该突变可能具有破坏性,评分为1分(0~1分),敏感度:0;特异度:1。高特异度低敏感度时,阳性诊断具有很大诊断意义。SIFT预测结果提示在POS 911从A到T的替代预计将影响ATP7B基因所表达的Copper-transporting ATPase 2蛋白质功能,得分为0<0.05,阳性。最后,明确该位点具有致病性与否,还需要结合其他位点的检测结果以及患者父母的检测结果进行进一步的分析。由于本例患者家庭条件困难,患者父母拒绝进行基因检测,这是个遗憾。
HLD虽然较为少见,但是在临床工作中如有遇到早发型锥体外系症状患者,建议把HLD作为需要考虑的鉴别诊断之一。本文旨在通过该例患者诊治及文献回顾,提高大家对HLD影像学特点以及致病基因(ATP7B)的认识,为以后对HLD患者的早期诊治提供帮助。

























