
强直性肌营养不良是一组以肌强直及肌无力为核心临床症状的遗传性肌病。往往因为个体临床表现和症状轻重程度极大差异,给临床诊断带来了一定的难度,所以标准化的诊断思路及方法是需要引起临床医生的关注。可以通过结合患者的肌肉系统外症状一定程度来减少临床医师的漏诊率。随着肌肉活体组织检查技术的推广及基因检测技术的改进,通过肌电图、病理及基因等辅助检查,诊断不存在困难。目前国际上治疗仍然以症状性治疗及症状管理为主。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
强直性肌营养不良(myotonic dystrophy,DM)是一类常见的常染色体显性遗传性肌病,其患病率约为12/100 000。DM多表现为进展性肌无力、肌强直及骨骼肌系统以外的多器官受损[1,2]。
DM包括1型和2型。DM1型是由于DM激酶(myotonin protein,DMPK)基因CTG序列扩增[3],而DM2型则是由细胞核酸结合蛋白(cellular nucleic acid-binding protein,CNBP)基因中CCTG重复扩增引发[4]。DM1型根据临床表型可分为:成人型、先天性和儿童型,其中以成人型最为常见[5]。DM2型则没有明显的临床亚型,多数为成人型。
DM1型的致病基因位于染色体19q13.2。其发病是由于DMPK基因的非编码区CTG三核苷酸重复序列异常扩增所致[3]。在正常人群中CTG重复扩增5~40次,而在DM1型患者中,其扩增数可超过100次。DMPK蛋白在心肌、骨骼肌中均有表达,也在脑组织中表达。CTG异常扩增影响RNA转录、剪接,这种毒性RNA进而影响骨骼肌氯离子通道功能,产生相应症状。
DM2型的致病基因位于染色体3q21.3,是由于锌指蛋白9(zinc finger protein 9,ZNF9)或称CNBP基因第1内含子CCTG核苷酸序列异常扩增所致[4]。mRNA异常剪接的离子通道蛋白使得离子通道功能下降,产生肌强直及无力等症状。目前认为在DM当中,毒性RNA是其主要致病因素,也是目前基因靶向治疗的热点。
DM患者中男女均可发病,多于青中年后起病。DM的临床表现主要包括肌肉症状和非肌肉症状。成人型DM1型是其最常见的类型。不同类型的DM临床症状差异较大(表1)。以下将着重描述成人型DM1型的临床症状。

强直性肌营养不良1型与强直性肌营养不良2型的临床特征[4]
Clinical manifestations in the myotonic dystrophy type 1 and myotonic dystrophy type 2[4]
| 项目 | 成人型DM1型 | DM2型 |
|---|---|---|
| 遗传方式 | 常染色体显性遗传 | 常染色体显性遗传 |
| 染色体 | 19q13.3 | 3q21.3 |
| 基因 | DMPK | CNBP |
| 核苷酸重复序列 | (CTG)n | (CCTG)n |
| 扩增部位 | 3 ′端非编码区 | 1号内含子 |
| 肌强直症状 | 常见 | 少于50% |
| 强直样肌电图 | 常见 | 不典型 |
| 无力症状 | 青中年期出现明显症状 | 中年老期可有无力症状 |
| 白内障 | 常见 | 少见 |
| 面肌受累 | 常见 | 罕见 |
| 眼睑下垂 | 常见 | 少见 |
| 吞咽困难 | 常见(疾病后期) | 罕见 |
| 呼吸肌受累 | 常见(疾病后期) | 罕见 |
| 近端肌肉受累 | 受累较晚 | 指深屈肌受累 |
| 远端肌肉受累 | 常见 | 常见,后期受累 |
| 胸锁乳突肌受累 | 常见 | 少见 |
| 肌痛 | 偶见 | 多见 |
| 肌萎缩 | 面肌、远端手部肌肉、下肢肌肉 | 罕见 |
| 肌酸激酶 | 正常、轻度或中度升高 | 正常、轻度或中度升高 |
| 心脏受累 | 常见 | 变异大,可严重影响心脏 |
| 肌颤动 | 罕见 | 明显 |
| 精神行为异常 | 常见 | 罕见 |
| 嗜睡 | 常见 | 少见 |
| 秃顶 | 常见 | 偶见 |
| 预期生存寿命 | 缩减 | 正常 |
注:DM:强直性肌营养不良;DMPK:强直性肌营养不良激酶;CNBP:细胞核酸结合蛋白
肌肉的核心症状为肌无力及肌强直。成人型DM1型可影响面肌、躯干肌、四肢肌、心肌、呼吸肌,出现肌无力、肌萎缩及肌强直症状。初期即可累及四肢远端(渐至近端)、肩胛肌、骨盆带肌,产生类似肢带型肌营养不良样症状,病情进展慢,症状轻,严重者最终也可导致严重残疾。肌强直多表现为用力握拳后松开困难,叩诊后肌球现象。疾病后期由于吞咽困难等症状,易合并肺炎等并发症。DM1型易累及颌面部肌肉。面肌、咀嚼肌的受累萎缩是DM1型的临床特征之一。咀嚼肌、面肌、咬肌无力,合并面肌萎缩形成的刀削样面容,称为"斧状脸",成为DM1型的特征性面容[2]。
DM1型的非肌肉症状可累及心血管系统、眼部、神经系统、消化系统、内分泌系统等。
心脏受累是DM的一大特点。由于心脏传导系统及窦房结的纤维化,使得心血管系统受累,出现传导性心律失常、肥厚性心肌病等一系列心血管疾病。并且需要警惕患者出现早发的心源性猝死,此为DM患者的主要死亡原因之一[6]。患者还容易出现亚临床的心脏疾病,仅在体检中发现心电图改变。
白内障是DM1型的另一个常见症状。白内障为晶状体变性、混浊而引起的视物模糊,多好发于40岁以上人群,随着年龄增长而发病率升高。在DM患者中,白内障的患病率明显增高。特别是对于发病年龄小于50岁的白内障患者,也需要敏感地考虑到DM1型的可能[7]。
对于DM1型患者而言,其神经系统的损害可以是器质性的,也可以是功能性的。最常见的症状为情绪障碍,存在有淡漠、轻度认知功能下降、嗜睡等症状。而神经心理障碍以及肌肉症状影响了患者的受教育程度、社交活动能力以及工作能力等。在头颅MRI中,也可以出现脑萎缩、广泛脑白质损害等表现[8]。
在DM1患者中还有一些少见的症状可影响消化系统、内分泌系统及生殖系统。秃顶是男性DM1型患者的特殊面容。消化系统的常见症状为便秘、腹泻、失禁等。DM1型患者胆囊相关疾病发病率也明显增高。在内分泌系统和生殖系统等方面,患者也存在胰岛素抵抗、甲状腺功能减低、性功能减退、男性乳房发育(图1)、不孕、流产等少见情况。尽管少见,但是上皮细胞肿瘤等一系列肿瘤性疾病的发生也不容忽视。在DM1患者中,子宫内膜、卵巢、大脑和结肠系统的肿瘤发生率约为正常人群的7倍[9]。
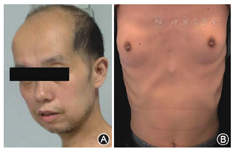
Clinical manifestations in the myotonic dystrophy type 1. A: Typical facial features in man: axe-shaped faces and early frontal balding; B: Gynecomastia in man
Clinical manifestations in the myotonic dystrophy type 1. A: Typical facial features in man: axe-shaped faces and early frontal balding; B: Gynecomastia in man
先天性DM1型患者在产前即可出现胎动减少、羊水增多,可在产前超声检查中发现有畸形。出生后表现为明显的肌张力降低,呈现为软婴,累及面肌、呼吸肌、躯干肌群、四肢肌肉,严重者可出现呼吸困难及喂养困难。与此同时,患儿还有不同程度的智力缺陷。
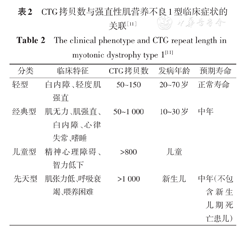
儿童型DM1型早期没有突出的肌无力、肌强直等临床表现,而更多是由于智力缺陷而被发现。随着年龄的增长,肌肉症状才逐步突显出来。对于没有家族史的患儿,早期易被漏诊[10]。一般认为CTG拷贝数越多,患者的临床症状越重,起病年龄越早(表2)。
DM2型患者相对而言临床表型差异较大,轻重不等,多为成年人,没有先天型和儿童型。对于轻者仅表现为持续性轻度肌酸肌酶升高,轻度肌肉无力,肌肉的僵硬、疼痛及疲劳感,可出现下肢近端无力等症状,导致爬楼困难,不累及面肌、呼吸、延髓肌。部分患者伴有肌痛,此类肌痛不易控制。部分症状较轻者可终生无明显症状,仅存在轻度不可用其他肝脏疾病等能够解释的轻度肌酸激酶升高。但是也有病情严重者,可在40岁出现明显的瘫痪致残,中年时期出现致死性呼吸衰竭、肌萎缩、心脏疾病等并发症,导致残疾,部分患者可出现心源性猝死。在DM2型患者中,心脏疾病相对于DM1型患者相对少见,但需要警惕冠心病的发生。肌强直症状的发生及电生理异常相较于DM1型患者少见。白内障等疾病的发病率也较DM1型低。DM2型罕有累及神经系统[12]。DM2型相对而言为良性病程,多数不影响患者的社会功能及预期寿命。
DM肌酸肌酶水平多呈轻度到中度升高,2型DM患者的肌酶水平甚至可以完全正常,同时可出现风湿学指标异常和血脂升高。由于强直性肌病患者同时存在内分泌、生殖系统疾病,所以还需要进行血糖、血脂、性激素等指标的检测。
包括心电图和肌电图。心脏损害是DM患者的主要非肌肉系统症状。心电图可以发现有宽PR间期,QRS波增宽,心房颤动及心房扑动等。在DM2型患者中,还可以观察到冠心病的心电图表现。DM患者的肌电图表现为:肌源性损害和强直电位,运动单位时限缩短,多相波增多。部分DM 2型患者的肌电图可表现正常(图2)。

Eelectromyography shows the combination of myotonia changes
Eelectromyography shows the combination of myotonia changes
主要包括头颅及肌肉的MRI。DM患者的肌肉MRI的最主要表现为肌肉脂肪化及肌萎缩,以下肢远端肌群受累为主。受累程度与患者临床症状相平行(图3)。在DM1型患者的头颅MRI中,可见有弥漫性脑白质病变,部分患者可出现脑萎缩。而在DM2型患者中,头颅MRI多表现正常。
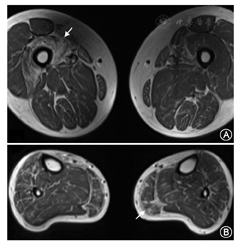
Magnetic resonance imaging of one myotonic dystrophy patient. A: Increased intramuscular T1 image signaling within the bilateral thigh muscles; B:T1-weighted hyperintense area in the musculi soleus
Magnetic resonance imaging of one myotonic dystrophy patient. A: Increased intramuscular T1 image signaling within the bilateral thigh muscles; B:T1-weighted hyperintense area in the musculi soleus
肌肉活检为DM的重要诊断方法之一。在DM患者的肌肉病理上,可以观察到明显的核内移、肌浆块、环形。肌纤维以及Ⅱ型肌纤维萎缩占主导(图4)。其中最典型的肌肉病理改变就是细胞核内移及肌浆块[13,14]。
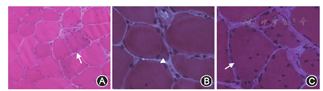
Differential histopathological features on muscle biopsy in the myotonic dystrophy patient. A: The hematoxylin and eosin-stained frozen section demonstrates the fibre size variation and an increased number of internal nuclei in the fibres ×20; B: The hematoxylin and eosin-stained frozen section demonstrates an increased number of nuclear clump fibres ×40; C: The hematoxylin and eosin-stained frozen section demonstrates an increased number of internal nuclei in the fibres ×40
Differential histopathological features on muscle biopsy in the myotonic dystrophy patient. A: The hematoxylin and eosin-stained frozen section demonstrates the fibre size variation and an increased number of internal nuclei in the fibres ×20; B: The hematoxylin and eosin-stained frozen section demonstrates an increased number of nuclear clump fibres ×40; C: The hematoxylin and eosin-stained frozen section demonstrates an increased number of internal nuclei in the fibres ×40
首先是心脏检查,DM患者的心电图可以呈现为传导性心律失常,心脏超声提示有心肌肥厚。在眼科检查的裂隙灯下,部分患者可以观察到晶状体变性、混浊。少数患者的子宫及双附件超声提示卵巢肿瘤性病变。部分患者的甲状腺超声可以出现甲状腺体积改变,部分回声降低,分布不均等。而在肠镜检测中,当患者有息肉、溃疡等表现时,需进行脱落细胞学检测有无肿瘤细胞。同时通过全身PET显像,发现肿瘤性病灶。
临床或者病理结果考虑有强直性肌病诊断的患者,需进行基因检测来进行确诊及分析。DM1型患者DMPK基因中CTG重复扩增数超过50次;DM2型患者CNBP基因中CCTG重复扩增数超过75次。
DM病情发展缓慢,自首发症状到最终确诊时间平均在5年以上,DM2型的临床症状不典型,病程时间更长,平均可达14年。所以临床上对疾病的识别显得尤为重要。由于当前电生理、病理及基因检测手段的日渐完善,依据肌强直及肌无力的临床表现,多系统受累(心脏、眼及内分泌系统)的症状和体征,结合肌电图、肌肉活检、基因检测等技术,本疾病的确诊不存在困难[2, 11]。DM患者的诊断可参考以下标准:(1)症状必须满足肌无力及肌强直的临床症状;(2)有不同程度的非肌肉系统等多器官受累的临床表现,白内障、心律失常是常见非肌肉系统症状;(3)"斧状脸""秃顶"是患者的特征性面容;(4)肌电图中典型的强直电位对疾病有明显的提示意义;(5)典型的肌肉病理改变为细胞核内移及肌浆块;(6)基因分析中存在有致病基因三核苷酸重复序列异常扩增。
对DM目前尚没有特异性的治疗方案。目前主要是对症治疗。首先应控制患者的肌强直症状,可给予美西律[2]。当发现患者有心律失常、冠心病等体征及症状时,应及时进行心血管科会诊,尽早干预。对于白内障患者,需要进行手术干预。值得注意的是,DM1型患者的麻醉风险较高,麻醉后呼吸恢复时间延长,易并发肺部感染等情况,而DM2型患者则没有麻醉风险增高的影响。
对于DM患者,需要监测其血糖、血脂及动脉硬化等情况。与此同时,对于高脂血症等患者,需要监测他汀类药物所产生的不良反应等。如出现明显的他汀药物所致的肌肉不良反应,应该及时停用他汀类药物。
目前虽然没有针对DM的特异性治疗,但是随着基因技术的进展,已经开展了基因靶向治疗的研究,特别是反义寡核苷酸治疗(antisense oligonucleotide,ASO)的研究。在实验动物模型中,通过注射ASO,已经可以达到通过降低RNA而逆转动物模型的生理、组织病理等改变的目的[8,11,15]。但是从动物实验到人体试验再到最终的临床应用依旧任重而道远。
DM病情进展缓慢,症状轻者可长期生活自理,甚至没有临床症状,仅有血清学轻度异常,多数不影响其生存寿命。部分重型患者,早期易发生心源性猝死[6],疾病后期由于心肺等并发症,可丧失工作生活能力,继发肺部感染及营养障碍导致死亡。
所有作者均声明不存在利益冲突
None declared
单选题(授予Ⅱ类学分说明及答题二维码见杂志内活插页)
1.下列有关强直性肌营养不良的描述中,错误的是?
A.强直性肌营养不良是最常见的肌营养不良症,男女均可发病,多于青中年后起病,分为1型及2型,其中以2型更为常见
B.强直性肌营养不良1型的致病基因位于染色体19q13.2。其发病是由于强直性肌营养不良激酶(myotonin protein,DMPK)基因的非编码区CTG三核苷酸重复序列异常扩增所致
C. DM2型的致病基因位于染色体3q21.3,2型是由于锌指蛋白9(zinc finger protein 9,ZNF9)基因第1内含子CCTG核苷酸序列异常扩增所致
D.强直性肌营养不良是一类常染色体显性遗传性肌病。多表现进展性肌无力、肌强直及骨骼肌系统以外多器官受损
2.男性患者,38岁,因“肌肉无力僵硬3年”于神经科门诊就诊。体格检查示:斧状脸,秃顶,上肢肌力Ⅴ级,下肢肌力Ⅳ级,大鱼际肌叩诊可见肌球。初诊时肌酸激酶500 U/L。下一步暂时不需要做的检查是?
A.肌电图、肌肉活检
B.基因检测
C.头颅MRI
D.心脏检查
E.骨SEPCT检查
3.男性患者,38岁,因“肌肉无力3年”于神经科门诊就诊。体格检查示:上肢肌力Ⅴ级,下肢肌力Ⅳ级,大鱼际肌叩诊可见肌球。初诊时肌电图可见强直电位,运动时限变窄,多相波增多。基因检测:DMPK基因的非编码区CTG扩增数大于100次。诊断考虑的疾病?
A.先天性肌强直
B.强直性肌营养不良
C. Isaacs综合征
D.高钾性周期性麻痹
4.强直性肌营养不良除了肌强直及肌无力以外,还有以下哪些表现?
A.胸腺瘤
B.肺癌
C.青光眼
D.白内障
5.关于强直性肌营养不良,下列哪项描述是正确的?
A.血清肌酸激酶和乳酸脱氢酶等肌酶水平显著增高
B.肌电图出现典型肌强直放电
C.肌活检显示大量的肌纤维坏死
D.首选治疗为基因靶向治疗

























