
肌萎缩侧索硬化(ALS)作为一种复杂的神经退行性疾病,其早期精准诊断十分困难。随着ALS相关遗传学的研究进展,越来越多的ALS致病及风险基因被发现,尤其是针对相关致病基因的基因治疗临床试验的开展,促进了基因诊断应用于ALS临床实践的必要性。本文介绍了目前常用的基因检测方法的优缺点,常见ALS致病基因的突变频率、基因型和表型的关系以及基因检测的策略。我国ALS患者最常见的致病基因是超氧化物歧化酶1(SOD1)基因,而在高加索人群中最常见的致病基因是C9ORF72基因。针对家族性ALS患者应建议其进行ALS致病基因panel的筛查,如果检测结果为阴性,需要行全外显子或者全基因组检测以寻找新的致病基因;对于散发性ALS患者,SOD1基因和C9ORF72基因均应作为常规筛查;此外,如果患者具有特殊的临床表型,病情进展迅速、病程极慢、合并认知损害、锥体外系症状等,为了进一步明确诊断和判断预后亦应进行相应的基因检测。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肌萎缩侧索硬化(ALS)是一类严重致死性的神经退行性疾病,目前尚无有效的诊疗手段[1]。分子遗传学研究发现遗传因素在ALS疾病发生发展中起了重要作用。近年来随着基因检测技术的不断提升,遗传学因素在ALS疾病发生发展中的作用逐步被解码,越来越多的ALS相关致病基因及风险位点被发现。尤其是近来开展的针对ALS致病基因的靶向治疗,使得ALS患者的预后发生了巨大变化,因此基因诊断将成为ALS早期精准诊断的有效手段,并为ALS的预后判断及基因治疗提供支撑。故此,本文主要介绍ALS常见的致病基因和由致病基因突变所致的临床特征。
基因诊断又称DNA诊断或分子诊断,是指通过准确、快速地获取生物体的遗传信息,从而对疾病做出判断的有效方法。目前,基因诊断已广泛用于感染性疾病的病原诊断、各种肿瘤的生物学特性的判断、遗传病的诊断等。随着越来越多的ALS致病基因及风险位点被发现,基因诊断也逐步被应用于ALS[2]。
基因检测技术是基因诊断的基础。随着分子诊断的快速发展,基因检测技术从时间长、成本高、通量低、大量手动操作,逐步变得越来越快速、低成本、高通量、自动化[3]。目前,基因测序技术已经成为检测基因变异的主要手段之一,包括第一代测序技术(Sanger测序)[4]、第二代测序技术(next generation sequencing)[5]和第三代测序[6]。
第一代测序技术通量小,每次测序仅可检测1 000 bp左右的片段;但测序准确度高,通常被用作标准的变异鉴定方法,来最终确定确切的变异位点和性质。第二代测序技术也被称作高通量测序技术,包括Panel测序、全外显子测序及全基因组测序等,其具有高通量、高准确性、高灵敏度、自动化程度高和低运行成本等突出优势。二代测序技术的发明,为大规模的全基因组范围的测序提供了可能。然而二代测序技术工作量大、费用高、读长较短,对一些大的结构变异无法识别。第三代测序技术是指在单个细胞、单分子水平上对基因组进行测序的一项新技术。第三代测序技术成功的克服了二代测序技术存在的诸多问题,具有测序通量更高,读取长度更长,测序时间更短等一系列优点[3]。
三种测序技术各有优缺点,需依据测序目的,合理选择最佳的测序方法,既可获得最佳的检测结果,亦节约成本。对于家族性ALS(fALS),可先针对常见的致病基因及风险位点进行Panel测序,若检测到可疑位点,可对患者及其直系亲属采用一代测序,对可疑位点进行验证;若检测结果呈阴性,可行基于全外显子或全基因组的二代测序,若检测结果仍为阴性,可考虑行三代测序。对于散发性ALS(sALS),尤其是早发性sALS以及一些临床表现异质性较大的sALS,可行全外显子测序以辅助疾病诊断。
自1993年,第一个ALS的致病基因超氧化物歧化酶1(superoxide dismutase,SOD1)基因被发现以来[7],越来越多的ALS相关基因被发现,尤其是近年来随着二代测序技术的普及,已经发现了百余个ALS相关基因[8],主要致病基因也达30余个(http://alsod.iop.kcl.ac.uk/)[2]。目前已证实的ALS致病基因主要以染色体显性遗传模式遗传,少数致病基因呈常染色体隐性遗传和X连锁显性遗传等[9]。据不完全估计,约68%的fALS和11%的sALS可被已证实的遗传因素所解释,其中最为常见的致病基因包括chromosome 9 open reading frame 72(C9ORF72)、SOD1、TAR DNA-binding protein(TARDBP)及fused in sarcoma(FUS)等[10]。不同的致病基因,甚至相同致病基因不同的致病位点可导致不同的临床表型,如起病年龄、临床症状以及疾病进展速度等具有显著差别。深入了解这些基因型对应的临床表型,有助于疾病的精准诊断、预后判断,并为今后的基因治疗提供依据。下面我们将重点介绍几种主要的ALS致病基因的基因型与临床表型的关系,并系统总结目前已证实的30余种致病基因可能的临床特点及致病机制(表1)。
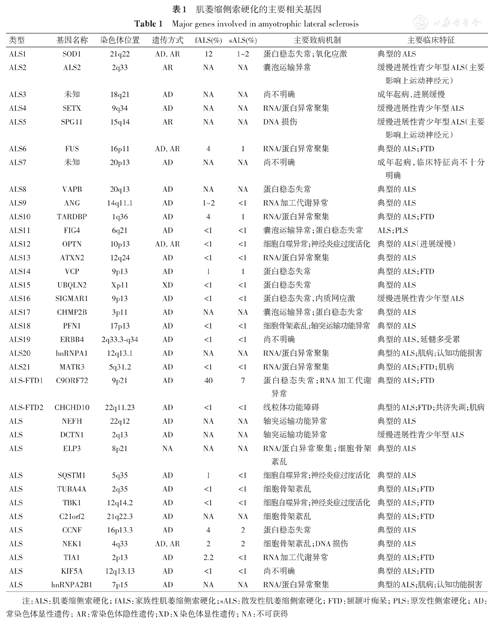
肌萎缩侧索硬化的主要相关基因
Major genes involved in amyotrophic lateral sclerosis
肌萎缩侧索硬化的主要相关基因
Major genes involved in amyotrophic lateral sclerosis
| 类型 | 基因名称 | 染色体位置 | 遗传方式 | fALS (%) | sALS (%) | 主要致病机制 | 主要临床特征 |
|---|---|---|---|---|---|---|---|
| ALS1 | SOD1 | 21q22 | AD, AR | 12 | 1~2 | 蛋白稳态失常;氧化应激 | 典型的ALS |
| ALS2 | ALS2 | 2q33 | AR | NA | NA | 囊泡运输异常 | 缓慢进展性青少年型ALS(主要影响上运动神经元) |
| ALS3 | 未知 | 18q21 | AD | NA | NA | 尚不明确 | 成年起病,进展缓慢 |
| ALS4 | SETX | 9q34 | AD | NA | NA | RNA/蛋白异常聚集 | 缓慢进展性青少年型ALS |
| ALS5 | SPG11 | 15q14 | AR | NA | NA | DNA损伤 | 缓慢进展性青少年型ALS(主要影响上运动神经元) |
| ALS6 | FUS | 16p11 | AD, AR | 4 | 1 | RNA/蛋白异常聚集 | 典型的ALS;FTD |
| ALS7 | 未知 | 20p13 | AD | NA | NA | 尚不明确 | 成年起病,临床特征尚不十分明确 |
| ALS8 | VAPB | 20q13 | AD | NA | NA | 蛋白稳态失常 | 典型的ALS |
| ALS9 | ANG | 14q11.1 | AD | 1~2 | <1 | RNA加工代谢异常 | 典型的ALS |
| ALS10 | TARDBP | 1q36 | AD | 4 | 1 | RNA/蛋白异常聚集 | 典型的ALS;FTD |
| ALS11 | FIG4 | 6q21 | AD | <1 | <1 | 囊泡运输异常;蛋白稳态失常 | ALS;PLS |
| ALS12 | OPTN | 10p13 | AD, AR | <1 | <1 | 细胞自噬异常;神经炎症过度活化 | 典型的ALS(进展缓慢) |
| ALS13 | ATXN2 | 12q24 | AD | <1 | <1 | RNA/蛋白异常聚集 | 典型的ALS |
| ALS14 | VCP | 9p13 | AD | 1 | 1 | 蛋白稳态失常 | 典型的ALS;FTD |
| ALS15 | UBQLN2 | Xp11 | XD | <1 | <1 | 蛋白稳态失常 | 典型的ALS |
| ALS16 | SIGMAR1 | 9p13 | AD | <1 | <1 | 蛋白稳态失常,内质网应激 | 缓慢进展性青少年型ALS |
| ALS17 | CHMP2B | 3p11 | AD | NA | NA | 囊泡运输异常;蛋白稳态失常 | 典型的ALS |
| ALS18 | PFN1 | 17p13 | AD | <1 | <1 | 细胞骨架紊乱;轴突运输功能异常 | 典型的ALS |
| ALS19 | ERBB4 | 2q33.3-q34 | AD | <1 | <1 | 尚不明确 | 典型的ALS,延髓多受累 |
| ALS20 | hnRNPA1 | 12q13.1 | AD | NA | NA | RNA/蛋白异常聚集 | 典型的ALS;肌病;认知功能损害 |
| ALS21 | MATR3 | 5q31.2 | AD | <1 | <1 | RNA/蛋白异常聚集 | 典型的ALS;FTD;肌病 |
| ALS-FTD1 | C9ORF72 | 9p21 | AD | 40 | 7 | 蛋白稳态失常;RNA加工代谢异常 | 典型的ALS;FTD |
| ALS-FTD2 | CHCHD10 | 22q11.23 | AD | <1 | <1 | 线粒体功能障碍 | 典型的ALS;FTD;共济失调;肌病 |
| ALS | NEFH | 22q12 | AD | NA | NA | 轴突运输功能异常 | 典型的ALS |
| ALS | DCTN1 | 2q13 | AD | NA | NA | 轴突运输功能异常 | 缓慢进展性青少年型ALS |
| ALS | ELP3 | 8p21 | NA | NA | NA | RNA/蛋白异常聚集;细胞骨架紊乱 | 典型的ALS |
| ALS | SQSTM1 | 5q35 | AD | 1 | <1 | 细胞自噬异常;神经炎症过度活化 | 典型的ALS |
| ALS | TUBA4A | 2q35 | AD | <1 | <1 | 细胞骨架紊乱 | 典型的ALS;FTD |
| ALS | TBK1 | 12q14.2 | AD | <1 | <1 | 细胞自噬异常;神经炎症过度活化 | 典型的ALS;FTD |
| ALS | C21orf2 | 21q22.3 | AD | NA | NA | 细胞骨架紊乱 | 典型的ALS;FTD |
| ALS | CCNF | 16p13.3 | AD | 4 | 2 | 蛋白稳态失常 | 典型的ALS |
| ALS | NEK1 | 4q33 | AD, AR | 2 | 2 | 细胞骨架紊乱;DNA损伤 | 典型的ALS |
| ALS | TIA1 | 2p13 | AD | 2.2 | <1 | RNA加工代谢异常 | 典型的ALS;FTD |
| ALS | KIF5A | 12q13.13 | AD | <1 | <1 | 尚不明确 | 典型的ALS;FTD |
| ALS | hnRNPA2B1 | 7p15 | AD | NA | NA | RNA/蛋白异常聚集 | 典型的ALS;肌病;认知功能损害 |
注:ALS:肌萎缩侧索硬化;fALS:家族性肌萎缩侧索硬化;sALS:散发性肌萎缩侧索硬化;FTD:额颞叶痴呆;PLS:原发性侧索硬化;AD:常染色体显性遗传;AR:常染色体隐性遗传;XD:X染色体显性遗传;NA:不可获得
SOD1基因编码超氧化物歧化酶1,是细胞内清除自由基和抗氧化的重要酶。ALS患者SOD1基因的突变分布于整个编码区,已有超过170种致病性突变报道[11],不同人种的突变位点具有一定异质性。在全球范围内,p.D90A是最为常见的突变类型;p.A4V是北美地区最常见的SOD1突变,约占全部SOD1突变的40%,但p.A4V突变在亚洲十分罕见[12]。大多数SOD1突变呈常染色体显性致病,但p.D90A突变在斯堪的纳维亚人群中呈常染色体隐性致病[13]。在我国,fALS患者首先要考虑SOD1基因突变,因为约25%的fALS由SOD1基因突变所致;而在sALS中的SOD1突变频率低,仅为1%~2%,但SOD1仍是我国sALS人群最常见的致病基因[14],而且有些突变会引起特殊的表型,如起病年龄、临床特征及生存期等方面均存在较大的差异。如携带p.A4V、p.H43R、p.L84V、p.G85R、p.N86S和p.G93A等突变的患者,疾病往往进展迅速,平均生存期小于3年;而携带p.G93C、p.D90A或p.H46R等突变,患者以下运动神经元损害为主要表现,进展缓慢,生存期可达10年甚至更久[15],临床上常常会怀疑ALS的诊断,遇此特殊表型的患者需要进行基因检测证实。如患者携带D90A-纯合突变,p.E100K、p.E100G、p.A89V、p.L84F、p.L84V、p.D76V、p.H46R、p.G37R和p.G10V等杂合突变,则表现为经典型ALS。由于针对SOD1突变而设计的反义寡核苷酸(antisense oligonucleotides,ASO)正在进行临床试验,有望为SOD1突变的ALS患者带来福音。因此,对于ALS患者应进行该基因的筛查,尤其针对fALS和特殊表型的sALS。
FUS基因作为ALS的一种重要的致病基因,其突变约占fALS的4%和sALS的1%,目前已有超过60种与ALS相关的突变,其中p.R521C最常见[16]。FUS基因突变多呈常染色体显性遗传,仅p.H517Q及少数新生突变呈常染色体隐性遗传[17]。FUS基因突变可见于成年起病的ALS,也可见于青少年起病的ALS、ALS伴额颞叶痴呆(FTD),在单纯的FTD中较为罕见。不同FUS基因突变患者的临床异质性较大,但多数患者中年起病,以颈部和近端肌肉受累为重,进展迅速,很快累及呼吸肌,生存期短[18];但也有突变,如K510R,则相对惰性,患者病程可达6~8年[19]。此外,智能减退、帕金森样症状等也可见于少数FUS基因突变患者[20],因此临床上如果ALS患者出现了这些症状,导致诊断存在一定困难,应该筛查该基因是否存在突变。
TARDBP基因编码TDP-43蛋白,这一蛋白是ALS的重要病理沉积物的主要成分,尽管仅有4%的fALS和1%的sALS由TARDBP基因突变所致[21],该基因仍是ALS的重要致病基因。目前已有50种与ALS相关的突变报道。通常TARDBP基因突变患者中年起病,高加索人群ALS患者多从上肢起病,而亚洲ALS患者多从延髓起病,病程长短不一,总体生存期相对较长(平均生存期63个月)。携带TARDBP突变的ALS患者既可表现为经典型的ALS,也可表现为ALS-FTD或ALS伴锥体外系症状,如帕金森病[22,23],由于TARDBP基因突变患者病例数不多,基因型和临床表型的关系需大样本的研究进一步明确。
C9ORF72基因是目前被证实的高加索ALS人群中最为常见的致病基因[24,25],45%~50%的fALS和5%~10%的sALS患者可能是由该基因变异所致[8,26]。但在我国ALS人群中,C9ORF72基因突变频率远远低于欧美人群。我们前期的研究发现,在sALS患者中C9ORF72突变频率仅约0.87%[27],后续我们将国内的研究进行汇总发现,仅约5.98%的fALS患者及0.53%的sALS患者可能是由C9ORF72基因变异所致[14],进一步证实了东西方人种ALS患者存在较大的遗传异质性。引起ALS的C9ORF72突变主要是由位于2个转录起始位点之间的GGGGCC(G4C2)六核苷酸序列异常重复扩增所致,正常个体的G4C2序列大多重复2~5次,而患者的重复次数可达数百乃至数千次[28]。C9ORF72突变的ALS患者多以延髓起病,常伴有认知功能障碍,如ALS-FTD,疾病进展迅速,中位生存期相对较短。此外,C9ORF72突变也可表现为FTD,甚至原发性侧索硬化(PLS)、进行性脊肌萎缩(PMA)及经典型ALS,因此基因型与表型的关系目前仍存在一定的争议,有待进一步大样本的病例验证[29]。目前针对C9ORF72突变而设计的ASO正在进行临床试验,有望为携带该基因突变患者的成功治疗带来希望。因此,尽管我国ALS患者C9ORF72突变频率低,仍应进行该基因突变筛查,尤其是针对伴认知功能损害的ALS患者,均应进行C9ORF72突变的检测。
此外,目前已明确的ALS致病基因,还包括VAPB、ANG、FIG4、VCP、TBK1、NEK1、TIA1及KIF5A等(表1)。尽管这些基因已经被证实与ALS相关,但是尚缺乏多中心、大样本的研究探究基因型与临床表型的关系,有些基因致病机制也尚不十分明确,有待进一步研究。
尽管目前国内外对于是否对ALS患者及家属进行基因检测还存在一定的争议[30]。但是目前大量研究证实,不论是fALS,还是sALS,均存在一定的遗传学基础。因此,对ALS患者开展遗传学检测,不仅为ALS的早期诊断及预后判断提供一定的帮助,同时筛查发现相关基因突变的患者及亲属也为今后基因治疗能应用于早期患者奠定基础。
首先,对于fALS可先检测已知的ALS相关致病基因及风险位点,尤其是C9ORF72、SOD1、TARDBP、FUS等主要致病基因,一旦发现阳性结果,应对家系中全部直系亲属进行验证,并结合基因型与临床表型之间的关系(表1),明确疾病的诊断。此外,致病基因的筛查也有助于指导家系成员的产前诊断和优生优育。对于检测结果呈阴性的fALS患者,最好行全外显子或全基因组测序筛查,并且通过家系共分离等技术寻找家系可能的新的致病基因。这样的研究不仅有助于患者明确诊断,同时可推动ALS发病机制的研究。
此外,对于sALS,尤其是临床特征不典型的ALS及疑诊的ALS均可进行基因检测,结合患者的基因型与表型特征(表1),有望尽早明确疾病诊断。虽然目前仅有少部分sALS病例找到了明确的致病基因,但是基于人群的研究显示,sALS的遗传度可高达61%,大量缺失的遗传度有望进一步研究证实[31]。因此,更多的在sALS患者中开展基因检测,有望发现新的ALS疾病相关基因。
ALS作为一种复杂的疾病,基因检测报告的解读也十分复杂。对检测报告阴性的患者,也不能完全排除遗传因素在该患者中的作用,毕竟目前已知的致病基因及风险位点仍占少数,专业人员应该结合患者的临床症状综合考虑。
ALS的遗传特征十分复杂,且患者平均生存期较短,使得ALS遗传相关研究开展十分困难。目前已知的多数ALS相关基因突变频率较低,且存在一定的种族异质性,加之基因检测费用相对昂贵,因而ALS的基因诊断在临床工作中的常规开展有一定难度。但是,随着科技的不断进步,越来越多的ALS遗传信息被解码,尤其是基于基因治疗技术的不断进步,ALS基因诊断将会被越来越多地应用于临床,为患者的早期诊断、预后判断乃至治疗提供帮助。
作者声明不存在利益冲突
None declared
单选题(授予Ⅱ类学分说明及答题二维码见杂志内活插页)
1.肌萎缩侧索硬化的遗传方式不包含以下哪项?
A.常染色体显性遗传
B.常染色体隐性遗传
C.X染色体连锁遗传
D.Y染色体连锁遗传
2.目前在欧美人群中最为常见的肌萎缩侧索硬化的致病基因是?
A.SOD1
B.FUS
C.TARDBP
D.C9ORF72
3.中国肌萎缩侧索硬化患者最常见的致病基因是?
A.FUS
B.C9ORF72
C.SOD1
D.PFN1
4.下列肌萎缩侧索硬化致病基因中,通常不出现额颞叶痴呆表现的致病基因是?
A.TARDBP
B.C9ORF72
C.SOD1
D.VCP
5.下列肌萎缩侧索硬化的致病基因中,往往导致青少年起病的是?
A.ALS2
B.C9ORF72
C.SOD1
D.FUS

























