
脑淀粉样血管病是脑血管疾病的一种特殊类型,文中介绍了该病的发病机制、病理改变、影像学特征、生物学标志、临床表现、诊断标准和治疗中的注意事项,并重点关注了与临床有关的研究进展。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2021年从第2期开始共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。
脑淀粉样血管病(cerebral amyloid angiopathy,CAA)是指淀粉样蛋白β(amyloid β,Aβ)在脑膜和皮质小血管壁内的沉积,主要见于小动脉和毛细血管,偶可发生于小静脉。继发于Aβ沉积的病理改变既可以引起血管壁破裂,产生脑叶自发性出血、微出血、大脑凸面蛛网膜下腔出血(convexity subarachnoid hemorrhage,cSAH)及其演变形成的皮质表面含铁血黄素沉积(cortical superficial siderosis, cSS),也可以导致白质高信号(white matter hyperintensity, WMH)和皮质微梗死(cortical microinfarct, CMI)等缺血性改变[1, 2, 3]。不同于其他神经系统疾病,对CAA的认识开始于病理学家,直到CAA与脑叶出血的相关性被发现后,该病才得到临床神经科医生的重视和关注[4]。神经影像学技术的进步使得CAA的生前诊断成为可能,主要基于影像表现建立的“波士顿诊断标准”已得到充分验证并大大促进了CAA的临床研究(表1)[5],分子和功能影像学技术也为了解其发病机制提供了新途径[6],所有这些共同推进了对CAA认识的不断深入。

修订的脑淀粉样血管病波士顿诊断标准[5]
Revised Boston diagnostic criteria for cerebral amyloid angiopathy (CAA)[5]
修订的脑淀粉样血管病波士顿诊断标准[5]
Revised Boston diagnostic criteria for cerebral amyloid angiopathy (CAA)[5]
| 肯定的CAA(definite CAA) | |
充分的尸检证实: | |
脑叶的皮质或皮质-皮质下出血 | |
严重CAA血管病 | |
排除其他病变 | |
| 很可能的CAA并有病理支持(probable CAA with supporting pathology, pr-CAA-P) | |
临床资料和病理(血肿清除组织或脑活组织检查)证实: | |
脑叶的皮质或皮质-皮质下出血(包括脑出血、脑微出血或cSS) | |
组织标本中存在一定程度的CAA | |
排除其他病变 | |
| 很可能的CAA(probable CAA) | |
临床资料和MRI或CT证实: | |
限于脑叶的皮质或皮质-皮质下多发出血(脑出血、脑微出血),允许存在小脑出血,或者单发脑叶皮质或皮质-皮质下多发出血合并cSS(局灶或播散性) | |
年龄≥55岁 | |
排除其他原因导致的出血 | |
| 可能的CAA(possible CAA) | |
临床资料和MRI或CT证实: | |
单发脑叶皮质或皮质-皮质下出血或cSS(局灶或播散性) | |
年龄≥55岁 | |
排除其他原因导致的出血 | |
注:CAA:脑淀粉样血管病;cSS:皮质表面含铁血黄素沉积;MRI:磁共振成像;CT:电子计算机体层扫描
Aβ在脑膜和皮质小血管壁内沉积是CAA发病的核心机制,其原因尚未完全阐明,普遍认为Aβ的清除障碍导致其在血管壁异常沉积,并非Aβ产生过多[7, 8]。Aβ的清除途径包括跨血脑屏障转运、细胞吞噬、酶降解和血管旁引流[9]。尽管还没有确切数据定量评价不同途径在Aβ清除中所占的比重,血管旁引流可能占据主要地位。推测的血管旁引流途径有两条,第一条途径位于小动脉基膜内,Aβ沿与动脉血流相反的方向从脑内向脑外转移[10, 11];第二条途径是类淋巴途径,在该通路中,脑脊液沿动脉周围间隙进入脑实质内,经由星形胶质细胞足突水通道蛋白4(aquaporin 4,AQP4)的转运进入组织间液,组织间液向静脉方向流动,再经AQP4转运进入静脉周围间隙,引流离开脑组织,在完成液体交换的同时,带走脑组织中的代谢废物[11, 12]。由于Aβ的沉积主要发生在小动脉的管壁中,推测第一种血管旁引流途径所起的作用更大。Aβ在动脉壁的沉积降低血管旁引流途径的效能,造成Aβ沉积的进一步加重,形成一个自我加强的恶性循环。
散发性CAA的发病年龄通常较晚,根据修订的“波士顿标准”,“很可能的CAA(pr-CAA)”和“可能的CAA(po-CAA)”的诊断都要求患者年龄在55岁以上[5]。但也有少数早发、非遗传性CAA病例被报道,这些病例多数有尸源性硬脑膜移植的病史,提示Aβ可能存在类似朊蛋白的传播机制,即致病性蛋白种子持续扩增,在细胞间扩布乃至跨区域扩散[13, 14]。在尸源性硬脑膜移植或注射尸源性生长激素导致的医源性克-雅病患者中也发现有CAA的病理改变[15]。Aβ在部分血管中呈局灶性分布,提示Aβ在最初沉积部位不断积聚、扩展,似乎也支持这一假说。
CAA和阿尔茨海默病(Alzheimer′s disease,AD)是两种独立的疾病实体,但Aβ的异常沉积是CAA和AD的共同基础[16],CAA和AD的病理改变经常相互重叠,对比两者的病理改变有助于理解CAA的发病机制。AD患者中,Aβ在脑实质内沉积形成神经炎斑,主要成分为Aβ42,CAA中的Aβ主要是Aβ40。Aβ40和Aβ42分别在血管和脑实质内选择性沉积,可能与Aβ片段的溶解性有关,Aβ42溶解性低,倾向于在脑实质内沉积,而溶解性较高的Aβ40能够沿血管周围清除途径弥散并在血管壁内沉积[16]。
影响Aβ沉积的因素还包括年龄和载脂蛋白E(apolipoprotein E,ApoE)基因多态性。尸检资料显示,80~90岁的非痴呆人群CAA发生率为20%~40%,在痴呆人群中可达50%~60%[17]。随年龄增长基膜的组织成分发生改变,可能影响了Aβ的清除效率。
ApoE是与胆固醇运输有关的血清蛋白, ApoE基因是已知的唯一与散发性CAA易患性有关的基因,ApoE有ε2、ε3和ε4 3种类型,ApoE-ε3/ε3是普通人群中最常见的基因型[16, 18, 19]。ApoE-ε4是CAA和AD的共同易患基因,与CAA-Ⅰ型有关,促进Aβ在血管壁沉积,包括毛细血管,并呈现剂量依赖性,ApoE-ε4携带者随年龄增长进展至CAA的风险升高。ApoE-ε2与CAA-Ⅱ型有关,与Aβ沉积所致血管继发病理改变的严重性相关,是出血型CAA的危险因素,但对AD是保护性因素[16, 20]。有假说认为ApoE-ε4加速Aβ在血管壁内的沉积,而ApoE-ε2促进继发出血性病理改变的发生,包括血管劈裂、纤维素样坏死和微出血[2]。
淀粉样前体蛋白(amyloid protein precursor, APP)基因突变也会造成Aβ构象改变和异常沉积。APP编码区附近的基因突变通常增加Aβ42的绝对数量或相对比例,倾向于形成神经炎斑,与早发的AD有关[21],而编码区内或近邻编码区的突变导致显著的CAA临床和病理表现。例如遗传性脑出血伴淀粉样蛋白沉积荷兰型(hereditary cerebral hemorrhage with amyloidosis-Dutch type,HCHWA-D),其基因改变位于APP基因的693号密码子内,导致软脑膜和皮质严重的CAA以及脑实质弥漫性Aβ沉积,但较少形成致密的神经炎斑,表现早发、复发性脑出血和痴呆,因其与散发性CAA在分子机制和临床表型的高度一致性,HCHWA-D为散发性CAA的研究提供了很好的模型[16, 22]。
脑膜和皮质小血管壁内的Aβ沉积是CAA的标志性病理改变。从全脑的范围看,Aβ沉积倾向于累及大脑半球后部,特别是枕叶、颞叶和顶叶,随病程进展可向前部扩展,偶尔累及小脑[23]。Aβ在血管壁的沉积可以有不同模式,从Aβ与血管基膜共存,到平滑肌细胞部分丢失,基膜成分仍有保留,直至血管壁的全部成分被Aβ所替代。在轻微CAA,仅有一小部分软脑膜和皮质浅表血管受累;在严重CAA,多数小动脉、微动脉均出现明显的Aβ沉积,中等大小的软脑膜动脉Aβ沉积出现在中外膜的外侧区域,小动脉和微动脉往往全层被Aβ所替代。但即使在严重CAA患者中,不同程度的Aβ沉积仍可呈斑片样交叉共存。在同一血管,病变严重的节段血管壁全层已被Aβ所替代,而紧邻节段可完全没有或仅有轻微的Aβ沉积[1, 24]。根据受累血管的类型,CAA可分为两型,当Aβ主要沉积于毛细血管时称为CAA-Ⅰ型,而没有毛细血管受累的称为CAA-Ⅱ型[20]。伴随Aβ的沉积,小动脉中膜平滑肌细胞进行性丢失,继发纤维素样坏死,血管管壁增厚,管腔狭窄,血管壁发生同心圆性劈裂,呈现典型的“血管内血管”改变,管壁薄弱处微血管瘤形成,最终引起脑组织的出血性或缺血性改变[24]。
CAA是自发性脑出血的主要原因之一,特别是在高龄患者中[25],除了脑出血,CAA导致的出血性改变更多以脑微出血的形式出现。CAA相关的脑出血或微出血位于脑叶表浅部位,倾向于累及后部颞叶和枕叶,与Aβ沉积的部位选择性一致[6],在脑微出血数量较多的患者中,病灶有在同一脑叶聚集的趋势(图1)[23]。而高血压性脑出血和微出血主要累及深部灰质和脑干,脑叶出血和微出血也是生前诊断CAA的主要线索[6]。
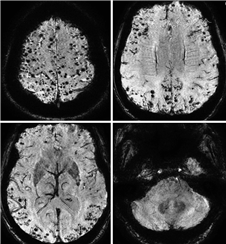
CAA相关脑出血容易向蛛网膜下腔扩散,边界不规则[26]。有研究结果显示,脑叶出血扩散至蛛网膜下腔、血肿边界手指样延伸和ApoE-ε4三者联合对于诊断CAA相关脑出血具有很高的敏感度和特异度,但研究样本量较小,且患者年龄偏大,尚需外部验证[27]。
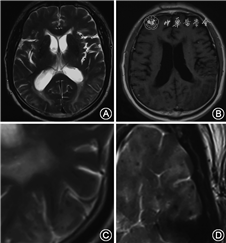
明显的脑微出血偶尔可在T2WI上看到(图2),但脑微出血的识别更多地依赖MRI梯度回波序列(gradient echo, GRE)或磁敏感加权序列(susceptibility weighted imaging, SWI)。微小血管病变导致血液外渗,在GRE或SWI序列上表现为2~5 mm大小的圆形低信号区(图1,3)。钙化、离子沉积、海绵状血管瘤等与脑微出血表现类似,应注意鉴别[28]。
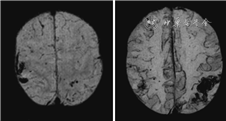
脑微出血和脑出血可能是既相互关联又相对独立的病理结局,并不是简单地代表了轻微和严重的CAA。一方面,基线脑微出血的负荷与新发脑出血的风险有关,新发出血的位置与基线脑微出血的位置也有一致性[23],体现了两者的内在联系。但也有研究结果显示在pr-CAA和po-CAA中,出血性病变的累加体积呈现双峰分布的特点,在尸检证实的CAA中,脑微出血较少(<3个)患者血管壁的厚度显著大于脑微出血较多者(>50个)[29]。
cSS指含铁血黄素在蛛网膜下腔、软脑膜和浅表皮质的沉积,在GRE或SWI序列上呈现沿脑沟分布的线样低信号(图3),有时在T2WI也可看到(图4)。如范围不超过3个脑沟,称为局灶性cSS,3个脑沟以上则称为播散性cSS,推测cSS最可能由cSAH所致[6, 30]。cSS对CAA的诊断具有较高特异度,在病理确诊的一组CAA(多数伴有脑出血)病例中其发生率可达60.5%,在经病理排除CAA的高血压脑出血对照组中则不存在[31]。在另一组pr-CAA病例中,cSS的发生率为40%,而在深部脑出血对照组中小于5%[32]。在鉴别脑出血和常染色体显性遗传脑动脉病伴皮质下梗死和白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy)时,cSS是唯一有价值且对CAA高度特异的影像学标志[33]。在修订的“波士顿诊断标准”中,cSS、脑叶出血和微出血对CAA的诊断被赋予了同等意义[5]。cSS还是预测CAA未来脑出血风险的可靠指标,并且cSS的范围越大,脑出血的风险越高[34, 35, 36]。
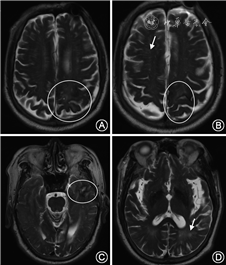
CAA患者出现WMH的原因与Aβ沉积导致的血管狭窄、继发缺血和血脑屏障的破坏有关。严重CAA患者WMH负荷重于健康老年人和AD患者。WMH对CAA并没有特异性,但WMH位置分布对其病因有一定提示意义。CAA导致的WMH常见于皮质下,而基底节周围的WMH更多与高血压性小血管病变相关[37, 38]。额叶或前后对称分布的WMH在健康老年人很常见,而后部优势的WMH常见于CAA(图4)[37, 39]。WMH导致不同脑区的连接中断,突出表现为执行功能损害和加工速率下降。CAA中WMH是认知功能损害的独立危险因素。伴随WMH负荷的增加,CAA性脑出血患者基线血肿的体积更大,血肿扩展和出血复发的风险也随之升高[6, 40, 41]。
扩大的PVS在MRI上可见,因此也常称为MRI可见的PVS,其信号特征与脑脊液类似,T2WI呈高信号、T1WI低信号,T2FLAIR示病灶周边无高信号,是与腔隙性梗死区别的关键[6, 28]。位置分布特征也是区分CAA和高血压性相关PVS的主要依据。前者主要位于半卵圆中心(图2),后者的PVS主要见于基底节区。半卵圆中心PVS的扩大可能提示Aβ清除系统的功能障碍,促进Aβ在脑膜和皮质表浅部位血管壁内沉积[42]。
CMI指位于皮质内直径小于5 mm的微小梗死,借助3 T或7 T高分辨MRI可以识别。与AD和健康对照相比,CMI更多见于CAA患者,并伴有较重的Aβ沉积负荷。CMI与脑微出血都是严重CAA的影像学标志[6]。影像可见的微小弥散加权成像(diffusion weighted imaging,DWI)高信号可能仅反映了CMI的冰山一角,一组生前存在脑叶出血和静息性DWI高信号患者死后的病理检查显示,一个DWI高信号估计与每年数百个新发的CMI相对应[43]。
CAA可伴有皮质萎缩,枕叶、颞叶、顶叶后部和颞叶内侧比较明显,与Aβ沉积较重的区域相对应。散发CAA和HCHWA-D患者都可出现明显的皮质萎缩,提示CAA可独立导致脑萎缩,并不依赖于散发CAA所伴发的AD病理改变[6, 44]。
MRI弥散张量成像能够重建白质传导束,提供全脑网络的信息[6]。与健康老年人相比,CAA患者脑网络连接效能下降,主要是与枕叶和颞叶后部联络的纤维损害,损害程度与Aβ负荷相关[45]。在随访中,CAA的严重程度与脑网络连接效能的恶化速度相关,连接性的下降导致执行功能障碍、加工速率下降和步态异常[45, 46]。
功能影像能够了解脑血管的功能改变。Aβ在血管壁的沉积导致内皮功能损害、血脑屏障通透性增加,血管自动调节能力受损,脑血管顺应性和对生理刺激的反应性下降[1]。评价CAA脑血管反应性的常用方法是利用功能MRI测定血氧水平依赖(blood oxygen level depending,BOLD)信号对视觉刺激的反应[6]。与健康老年人相比,CAA患者接受视觉刺激后BOLD信号变化的幅度降低,反应时间延长[47]。BOLD信号降低的幅度呈阶梯状空间分布,在枕叶最为明显,并与脑微出血和WMH负荷相关[48]。在随访中,BOLD信号的反应呈进行性下降趋势[49]。
分子影像通过标记致病蛋白可以反映CAA的发病机制、功能状态和病理改变。用于标记Aβ的常用标志物是11C标记的匹兹堡复合物B(11C-Pittursburgh complex B,PiB),能够与血管和脑实质内的Aβ结合[6, 50]。相对于非痴呆的健康老年人和高血压性脑出血患者,CAA患者全脑对PiB的摄取增加[51]。从定性评价的角度,pr-CAA患者中PiB阳性率可达77%~92%,PiB阴性基本可以除外CAA,但PiB阳性对CAA的特异度相对较低(66%~88%),可能与亚临床AD或增龄相关的Aβ沉积有关[50, 52]。
Aβ沉积是CAA和AD共同的分子发病机制,通过PiB摄取的定量或定性评价鉴别CAA与早期AD非常困难。由于CAA在枕叶更为严重,而AD额叶病变突出,多不累及枕叶,不同部位PiB摄取的差异在CAA和AD的鉴别中曾被寄于厚望,的确也有研究发现CAA患者枕叶与全脑皮质PiB摄取的比值总体上高于AD患者,但CAA和AD个体间的结果存在重叠交叉,并且有额叶高摄取、枕叶低摄取的病例最终经病理诊断为CAA[53, 54]。总体看来,尽管CAA患者的确存在从前向后的PiB摄取阶梯,枕叶与全脑皮质PiB摄取比值在区分CAA和AD中的意义仍不明确[50]。
分子影像是唯一能够显示在体Aβ沉积的技术,也为了解CAA的病理生理机制提供了新方法。比如,在AD或皮质下血管性认知功能障碍患者中,cSS和cSAH全部出现在全脑PiB摄取增高的患者;在CAA患者中,全脑PiB摄取水平与WMH体积、半卵圆中心的PVS数量、全脑网络的完整性相关,而在AD患者中并未发现全脑PiB摄取与WMH的体积有关。从空间分布看,PiB摄取增高的位置与基线脑微出血、新发微出血和出血以及脑网络破坏的位置具有空间一致性[6, 50]。
Aβ清除障碍是CAA的主要发病机制,其脑脊液中的Aβ水平可能存在异常改变。CAA患者脑脊液Aβ40的含量低于健康对照和AD患者,而Aβ42的水平也低于健康对照,但和AD患者类似。症状性HCHWA-D和症状前HCHWA-D患者脑脊液Aβ40和Aβ42水平也低于健康对照[55, 56]。
CAA相关脑出血根据部位不同可出现相应的定位症状,并无特异性。
短暂局灶性神经发作(transient focal neurological episodes,TFNEs)是脑叶出血之后CAA第二常见的临床表现。TFNEs可类似短暂性脑缺血发作(transient ischemic attack,TIA)、有先兆偏头痛和癫痫。典型的TFNEs表现为反复、快速、刻板的神经症状,如感觉异常、无力、语言障碍等,持续时间通常不超过30 min[57, 58]。TFNEs与cSS、cSAH和脑微出血密切相关,在416例诊断高危TIA和小卒中患者中,3.1%的患者存在cSS或脑叶微出血[59]。在CAA合并TFNEs的患者中,cSS的发生率达50%,而在没有TFNEs的患者中仅为19%[30]。越来越多的证据显示,TFNEs是急性cSAH的结果,其机制尚不确定,可能通过皮质扩布性抑制或痫性发作介导[60]。
慢性进行性认知功能损害是CAA的另一常见表现[1, 57, 60]。一组无痴呆、不伴脑出血的po-CAA患者在随访1年后痴呆的累积发生率为14%,5年时达到了73%[61]。出血前没有痴呆的一组脑出血患者1年后痴呆的总体发生率为14.2%,而脑叶出血组达23.4%[62]。来自社区老年人的尸检资料显示,中重度CAA的病理改变与认知功能损害有关,并且预示全域认知功能下降的速度更快。对共存的出血和AD相关病理改变进行校正后,CAA仍是认知功能损害的独立危险因素[60]。
不同于AD患者突出的情景记忆损害,CAA的认知功能下降主要表现为加工速率下降、执行功能受损和注意力缺陷[1, 57, 60]。CAA的认知功能损害特点也可以类似其他经典的临床痴呆综合征,有表现原发性进行性失语症和路易体痴呆典型症状的病例最终经病理证实为CAA[63]。
尽管ICAA与CAA密切相关,但其临床特征显著不同于CAA。ICAA可分为CAA相关炎症(CAA related inflammation, CAA-ri)和Aβ相关血管炎(Aβ related angiitis, ABRA), 两者分别以血管周围炎性浸润和透壁性血管炎为病理特征。ICAA可发生于多个部位,仅有1/3累及枕叶[64, 65]。ICAA症状多样,以亚急性认知功能下降、癫痫和头痛最为常见[66]。ICAA通常为不对称的大片白质病变,影像学特点符合血管源性水肿的特征,可有占位效应、脑实质或脑膜强化(图4),与AD患者接受抗Aβ单克隆抗体治疗后出现的“Aβ相关影像异常(Aβ related imaging abnormality, ARIA)”类似,后者包括大片脑实质水肿、脑沟渗出和脑微出血[16, 65]。ICAA和ARIA可能具有类似的发病机制,针对血管壁内Aβ的免疫反应起到关键作用。在2篇个案报道中,ICAA和ABRA患者的脑脊液中均发现了针对Aβ的抗体[65]。相对于原发中枢神经系统血管炎,ABRA发病年龄相对较大、蛋白升高更为明显、较少发生梗死,出血发生率更高,脑膜强化更为常见,但均不具有特异性[67]。
激素和免疫抑制剂是治疗ICAA主要药物,接受治疗的患者多数有影像或症状改善,但缓解后复发并不罕见[66],长期免疫抑制治疗的必要性值得进一步探讨。
总体而言,目前还没有能够改变CAA自然病程的方法,CAA患者的管理主要关注于如何降低出血风险。
来自PROGRESS(the Perindopril Protection Against Recurrent Stroke Study)实验的亚组分析结果显示,在有脑出血病史的患者中,培哚普利降压治疗降低了脑出血复发,包括po-CAA相关(77%)、高血压性(46%)和未分类的脑出血(43%),但是po-CAA相关脑出血仅有16例[68]。一项单中心的脑出血队列观察性研究结果显示,血压控制不良与脑叶和非脑叶的出血复发均有关系[69],提示对CAA患者应严格控制血压于正常范围内。
SPARCL(Stroke Prevention by Aggressive Reduction in Cholesterol Levels)研究发现阿托伐他汀可能增加脑出血的风险[70],CAA患者能否接受他汀治疗也受到关注。但一项病例对照研究和荟萃分析结果显示,脑出血发生前使用他汀能够减轻90 d时的病死率和致残率[71]。另一观察性研究发现他汀主要与CAA背景下的脑出血相关[60]。来自意大利的多中心研究也提示他汀对未来出血风险的影响主要发生在脑叶出血患者中[72]。尽管他汀对脑出血的影响还不十分明确,脑叶出血或多发脑微出血患者选择他汀治疗时需要谨慎评估[60]。
相当数量的TIA、缺血性卒中以及心房颤动患者可能合并CAA,在这些患者中合理使用抗凝和抗血小板药物是一项颇具挑战性的任务。CAA相关脑出血的年复发率达9%左右,大约是高血压性脑出血的5倍[73, 74]。脑出血后生存的患者中,华法林使出血复发的风险又增加了5倍[75]。尽管有数据显示脑出血合并心房颤动的患者在出血后重新使用抗凝药能够降低病死率,但是这些研究均未区分CAA导致的脑出血和高血压性脑出血[60, 76]。一项应用决策分析模型进行的研究结果显示,在脑叶出血合并心房颤动的患者中应避免抗凝药的应用[65]。这一结论或许也适用于存在播散性cSS或cSAH的患者,因其同样提示未来出血的高风险[74]。根据欧洲心脏病协会2016的心房颤动指南的相关内容,在有脑叶出血史、pr-CAA或肯定的CAA(d-CAA)中应避免抗凝药物的应用[77]。在心房颤动伴高危出血风险的患者中,左心耳封堵也是一项可能的选择[74]。
偶然发现的脑叶微出血对抗凝出血风险的影响尚无定论,一项荟萃分析结果显示在缺血性卒中、心房颤动和脑微出血(包括脑叶和深部脑微出血)并存的患者中,华法林导致出血风险增加了4倍[78],而其中脑叶微出血患者的出血风险可能更高,脑叶多发微出血患者应尽可能避免抗凝治疗。
存在脑叶微出血的普通人群在平均4.9年的随访中,脑出血的年发生率仅为0.6%,TIA或缺血性卒中患者如果合并脑微出血会同时升高缺血性卒中和出血性卒中的风险,而缺血性卒中的绝对风险超过出血性卒中[79, 80],少量脑叶微出血合并缺血性卒中高风险的患者仍有从抗栓治疗中获益的可能,但当脑微出血超过5个的患者服用抗血小板药物时,这种获益可能被抵消[81]。当患者的CAA诊断主要依据脑微出血的数量和位置时,其脑微出血的数量基本在5个以上。除非存在极高的缺血性卒中风险,多数CAA患者可能需要避免抗血小板药物的应用。
作者声明不存在利益冲突
None declared
1.脑淀粉样血管病(CAA)中淀粉样蛋白β(Aβ)沉积最显著的部位是?
A. 基底节
B. 小脑
C. 枕叶
D. 额叶
2. 对于鉴别CAA和常染色体显性遗传动脉病伴皮质下梗死和白质脑病(CADSIL)有价值的影像学表现是?
A. 腔隙性梗死
B. 皮质表面含铁血黄素沉积
C. 脑微出血
D. 扩大的血管周围间隙
3. CAA患者中血管壁内沉积的蛋白主要是?
A. tau蛋白
B. Aβ42
C. Aβ40
D. α突触核蛋白
4. CAA患者应首先避免应用的药物是?
A. 抗凝药物
B. 抗血小板药物
C. 他汀类药物
D. 降压药物
5. 根据波士顿诊断标准诊断,诊断可能的CAA时患者年龄应?
A. ≥40岁
B. ≥50岁
C. ≥70岁
D. ≥55岁

























