
脑白质病伴共济失调是一种由CLCN2基因变异引起的极为罕见的常染色体隐性遗传病。常见临床表现包括小脑性共济失调、头痛、认知障碍等,其头颅磁共振改变具有明显特征,目前报道的病例较少。现报道1例我院近期收治的48岁女性脑白质病伴共济失调患者的临床资料,供临床诊治参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脑白质病伴共济失调(leukoencephalopathy with ataxia,LKPAT)是一种极为罕见的常染色体隐性遗传病,常见的临床表现包括小脑性共济失调、头痛、认知障碍、视觉异常等,其头颅磁共振成像(magnetic resonance imaging,MRI)改变具有明显特征,主要表现为双侧内囊后肢、大脑脚、小脑中脚对称性的信号异常[1]。该病是由CLCN2基因变异所致,现报道1例CLCN2基因变异所致的LKPAT患者的临床资料,并总结其临床和影像学特点,旨在为临床诊治该疾病提供参考。
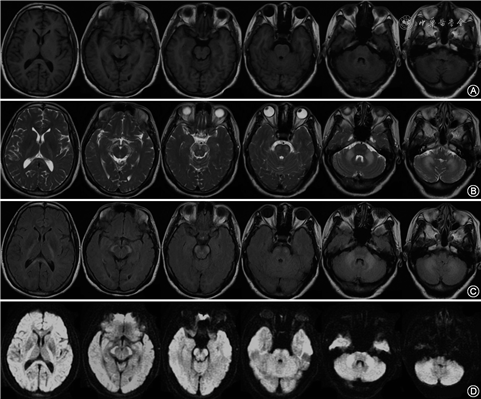
患者女性,48岁,以“发作性头痛10年余,行走不稳7年余”于2020年6月25日就诊于复旦大学附属华山医院。患者于2010年无明显诱因出现发作性头痛,主要表现为顶、枕部胀痛,伴恶心,无畏光和呕吐,一般休息2~3 d后好转,每年发作3~5次。2013年起患者开始出现行走不稳伴头晕和记忆力减退,稍复杂的家务完成有困难。症状呈进行性加重,无言语不清,无肢体麻木无力,无脚踩棉花感等表现。患者既往史无特殊,否认家族史和父母近亲婚配史。其父亲因“食管癌”在50岁时去世。神经系统体检:意识清楚,近记忆力、计算力、执行能力下降;简易精神状态检查量表评分为24分,蒙特利尔认知评估量表评分为18分。脑神经检查未见明显异常,四肢肌力Ⅴ肌,肌张力正常,四肢腱反射亢进(+++),双侧Babinski征(+);双侧深浅感觉对称无明显异常,双侧指鼻试验、跟膝胫试验欠稳准,一字步困难。入院后辅助检查:血常规:血红蛋白104 g/L(正常值115~150 g/L),肝肾功能、糖化血红蛋白、甲状腺功能、维生素B12、叶酸、同型半胱氨酸、梅毒血清学试验、HIV抗体、抗核抗体和肿瘤标志物均正常;2020年6月8日,患者行头颅MRI(图1)提示双侧内囊后肢、大脑脚、脑桥、小脑中脚及小脑对称性异常信号,T1WI呈低信号,T2WI、T2 FLAIR和DWI呈高信号。磁敏感加权成像(susceptibility weighted imaging)未见明显异常,磁共振波谱分析(magnetic resonance spectroscopy,MRS)提示N-乙酰天冬氨酸(N acetyl aspartic acid,NAA)峰未见明显降低,胆碱峰未见明显升高,胆碱/NAA比值在正常范围内。脑电图正常。
根据患者行走不稳症状及体检所见的小脑性共济失调体征和双侧锥体束征,结合头颅MRI,定位诊断为小脑和双侧锥体束病变。该患者为慢性进展性病程,头颅MRI提示双侧对称小脑、小脑中脚及锥体束受累,定性诊断首先考虑为遗传性脑白质病。结合其病灶在MRI上的特征性分布和改变,临床高度疑诊LKPAT,遂行遗传性脑白质病基因检测,结果发现CLCN2基因(在线人类孟德尔遗传数据库600570)存在纯合突变c.211C>T (p.R71*)(图2),为无义突变,参照美国医学遗传学与基因组学学会(the American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南[2],判定为可能致病性突变,由此该患者确诊为LKPAT。该病需与其他同时累及锥体束和小脑的遗传性脑白质病鉴别,如脑腱黄瘤病、成人多糖体病等[3, 4]。患者母亲基因检测发现CLCN2基因存在杂合突变c.211C>T(图2)。给予患者丁螺环酮 5 mg 3次/d口服,1个月后随访时患者自诉行走不稳改善,但认知障碍无明显变化。
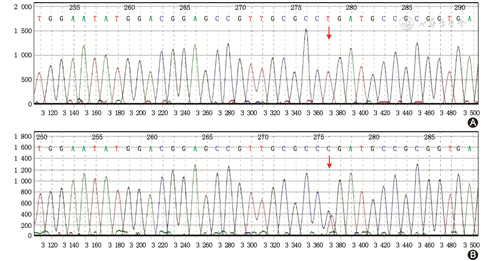
LKPAT是一种由CLCN2基因变异引起的罕见常染色体隐性遗传病。CLCN2基因定位于3号染色体长臂,编码氯离子通道蛋白2(chloride channel-2,ClC-2),其广泛表达于大脑、睾丸、结肠等26种组织中[5]。Depienne等[6]发现,ClC-2在维持脑组织的水电平衡中发挥重要作用。常染色体隐性CLCN2基因功能缺陷将影响脑组织的水电解质平衡,引起渗透性髓鞘内水肿,进而出现脑白质病。LKPAT由Depienne等[6]于2013年首次报道,Guo等[1]2019年报道了国内首例患者。目前全世界仅有14例患者报道,其中12例为CLCN2基因纯合突变,2例为CLCN2基因复合杂合突变[1,6, 7, 8, 9, 10, 11]。本例患者经基因检测发现3号染色体184076772位点发生突变,对应CLCN2基因NM_004366转录本的211位核苷酸发生C>T的纯合突变,导致其编码的71位氨基酸的终止密码提前出现,为无义突变。该变异所在区域是ClC-2蛋白的重要组成部分,不同物种的氨基酸序列高度保守,参照ACMG遗传变异分类标准与指南,判定为可能致病性突变。后续的家系验证发现,患者母亲存在CLCN2基因杂合突变c.211C>T,患者父亲因“食管癌”在50岁时去世,未能对其进行基因检测。我们推测,患者父亲亦存在CLCN2基因杂合突变c.211C>T。通过文献检索该突变未被报道。
在已报道的LKPAT病例中,以女性居多,平均发病年龄为27.8岁。常见的临床表现包括小脑性共济失调、头痛、动作性震颤、认知障碍、视觉异常和精子缺乏等,其中以共济失调最为常见[1]。该病进展缓慢,多数患者神经功能缺损症状轻微。目前尚无疾病相关失明、严重运动障碍及死亡的病例报道,故可认为是一种相对良性的遗传性疾病。本例患者的主要临床表现包括小脑性共济失调、头痛和认知障碍,与既往报道相符[1,6, 7, 8, 9, 10, 11]。
LKPAT特征性的MRI改变为双侧内囊后肢、大脑脚、小脑中脚的对称性的信号异常,T1WI低信号,T2WI、T2FLAIR及DWI高信号;也可累及脑桥锥体束、小脑上脚、胼胝体及其他大脑与小脑的白质,而脑室旁白质多不受累[1]。弥散张量纤维束成像可发现锥体束、桥横纤维和小脑齿状核纤维明显变薄[1]。该MRI改变具有显著特征性,是支持诊断的重要临床线索。Depienne等[6]认为CLCN2基因功能丧失导致脑组织水电失衡,引起渗透性髓鞘内水肿,是出现这种特征性MRI改变的重要原因。本例患者头颅MRI所示病变分布、范围以及不同序列的成像特点与既往报道相符。此外,我们还对本例患者进行了MRS检查,未发现NAA峰、胆碱峰及胆碱/NAA比值的异常,有待未来在更多的患者中进一步验证。
目前,LKPAT尚无特异性的治疗方案。丁螺环酮是一种5-羟色胺1A受体部分激动剂,常用于治疗焦虑症。有临床研究发现,丁螺环酮能改善橄榄桥小脑萎缩患者的共济失调[12]。本例患者使用丁螺环酮显著改善了共济失调,但其在LKPAT患者中的应用及疗效仍有待于更多的临床积累及探索。
我们报道了CLCN2基因纯合突变所致的LKPAT,在国内十分罕见,有助于国内神经内科医生提高对LKPAT的认识,拓展脑白质病的鉴别诊断谱,以免误诊和漏诊。该病极为罕见,临床症状多轻微且缺乏特异性,但MRI有特征性改变,是支持诊断的重要线索,最终确诊依赖于基因检测发现CLCN2的纯合或复合杂合突变。
所有作者均声明不存在利益冲突
None declared

























