
常染色体显性遗传脑动脉病伴皮质下梗死及白质脑病(CADASIL)是由NOTCH3基因突变导致的遗传性脑小血管病。主要临床表现为在疾病不同阶段出现偏头痛、缺血性脑卒中、进行性认知功能障碍、精神心理异常等,头颅磁共振成像可见多发腔隙性脑梗死、脑白质T2高信号及脑微出血。CADASIL的诊断“金标准”是病理检查发现微小动脉平滑肌细胞表面出现嗜锇性颗粒物质和(或)基因检查发现NOTCH3基因致病变异。本文重点介绍CADASIL的发病机制、临床表现、辅助检查、诊断和鉴别诊断以及疾病管理。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2021年从第2期开始共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。
尽管遗传性脑小血管病有多种类型,但常染色体显性遗传脑动脉病伴皮质下梗死及白质脑病(cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy,CADASIL)是最常见的遗传性脑小血管病,致病基因为NOTCH3基因,该病主要表现为在疾病不同阶段出现偏头痛、缺血性脑卒中、进行性认知障碍以及精神心理异常等,头颅磁共振成像(magnetic resonance imaging,MRI)检查可见脑白质T2高信号、多发腔隙性脑梗死及脑微出血。其诊断“金标准”有2个,分别为病理检查发现微小动脉平滑肌细胞表面出现嗜锇性颗粒物质(granular osmiophilic material,GOM)和基因检查发现NOTCH3基因致病变异。我国学者在2003年首次报道了通过基因检测和病理检查证实的CADASIL患者[1]。随着对该病认识的逐步加深和经验积累,我国CADASIL患者的临床特点也日渐明确。
NOTCH3基因编码的NOTCH3蛋白属于单次跨膜受体蛋白,包括胞外段、跨膜段和胞内段3部分。其中胞外段包含34个表皮生长因子样重复结构域(epidermal growth factor-like repeat,EGFr),每个EGFr结构域内有6个半胱氨酸残基,互相配对形成3个二硫键,构成EGFr结构域的次级结构。NOTCH3基因在成年个体中主要表达于血管系统,对血管平滑肌细胞的成熟和正常功能的维持起到重要作用。NOTCH3基因有33个外显子,95%以上的致病变异位于第2~24号外显子。目前全世界已报道了近300多种CADASIL致病变异,包括我国患者中发现的新变异位点。大部分致病变异为累及半胱氨酸残基的杂合错义突变,导致胞外段EGFr结构域内半胱氨酸残基的数目由偶数变为奇数,从而影响二硫键的配对;仅有少数致病变异不累及半胱氨酸残基。在高加索人群患者及我国北方患者中,第3、4号外显子是致病变异的热点区域,其次是第5、6、8、11、20号外显子。而在我国南方患者及韩国患者中,第11号外显子的p.R544C突变是热点突变[2]。
CADASIL主要累及全身微小动脉,在中枢神经系统主要累及小的穿支动脉和软脑膜动脉。电镜下可观察到血管平滑肌细胞变性、丢失,小动脉平滑肌细胞基底膜增厚和出现GOM结构。GOM通常呈直径1~2 μm的蘑菇状或不规则圆形,其底部与血管平滑肌细胞基底膜贴近,头部突向细胞外基质(图1)。毛细血管周细胞也可出现退行性改变,其表面也可出现GOM沉积。免疫电镜显示GOM中含有NOTCH3蛋白胞外段。
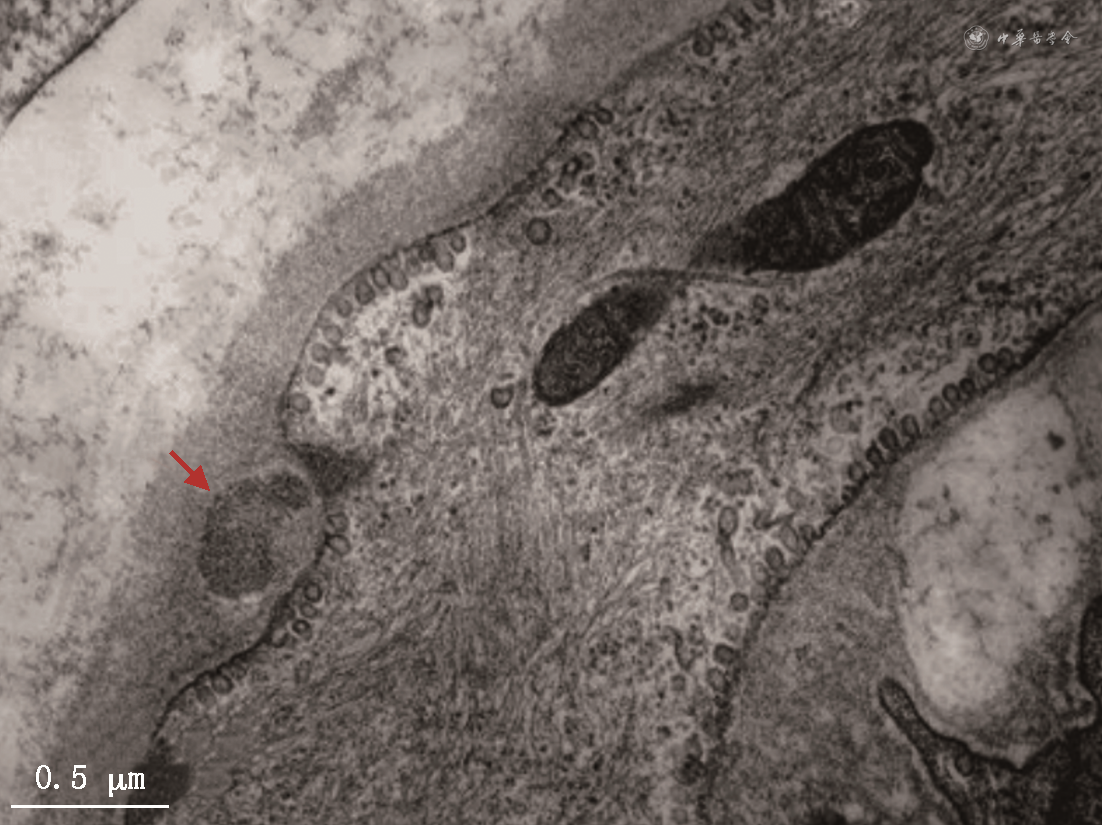
NOTCH3基因突变通过不同途径引起小动脉病变:一方面,基因突变导致其编码的NOTCH3蛋白获得新的毒性功能,通过自我聚集在细胞表面形成二聚体或多聚体,螯合在细胞外基质蛋白上,介导GOM的形成及并导致细胞基底膜增厚;另一方面,突变型NOTCH3蛋白还导致血管平滑肌细胞内关键分子通路失调、细胞骨架结构紊乱、膜离子通道异常、细胞增殖能力改变等[3]。综上所述,脑小动脉结构与功能异常,引起脑血流动力学障碍,造成脑组织低灌注,从而出现腔隙性脑梗死、白质脱髓鞘以及微出血、伴随扩大的血管周围间隙等病理改变[4]。
患者的起病年龄主要分布在30~60岁,主要表现如下。
在中国内地,偏头痛的发病率为17.2%[5]。偏头痛多在疾病早期出现,最早可于10岁之前出现,但发生偏头痛的平均年龄在30岁左右。患者可以表现为伴或不伴先兆的偏头痛。高加索人群中的偏头痛发病率较高,偏头痛的发生与患者疾病是否进展无关[6]。
短暂性脑缺血发作或缺血性卒中是CADASIL最常见的临床表现,出现在我国68.0%的患者当中,和日本的研究数据类似[5]。患者首次发生缺血性事件的年龄变异较大,平均发病年龄为40~50岁[7],表现为面部或肢体的麻木力弱、构音障碍、饮水呛咳等。患者在多次缺血性卒中后会遗留肌张力增高和肌力下降,导致独立行走能力逐渐下降,90%的患者在疾病晚期伴有步态异常[8]。心脑血管危险因素会加重患者的症状,收缩压升高是诱发脑梗死的危险因素之一,而吸烟、劳累、熬夜会促发缺血事件的发生。急性低血压也可以导致脑卒中的发生[9]。
约3%的CADASIL患者会出现脑出血,在基底节、丘脑和脑叶均可发生,也可以作为该病的首发症状出现,过多的脑微出血灶、控制不良的高血压、抗凝或抗血小板药物的应用可能会增加自发性脑出血的风险[10]。携带p.R544C位点突变的患者脑出血的发生率高于其他位点突变的患者[10]。
认知功能损害是该病的另一主要临床表现,出现在我国57.4%的CADASIL患者中[5]。大部分患者在疾病的最后阶段都会出现一定程度的认知功能损害,其认知功能改变规律与其他血管性痴呆类似,早期以执行功能减退和信息处理速度减慢为主,语言流畅性和注意力受损也较多见,随着疾病发展,一些患者会出现即刻和延迟回忆的异常,疾病晚期可出现各认知领域的全面损害[11]。认知功能损害是导致患者自理能力丧失的主要原因之一,与白质纤维束的广泛破坏以及皮质神经细胞损害有关[12]。低收缩压会加重患者脑血流动力学的异常,增加患者出现认知功能损害的风险[13]。
CADASIL患者的精神症状包括抑郁、焦虑、淡漠以及较为少见的双相情感障碍及精神分裂样症状等,且少数患者以情绪障碍为首发症状[14]。抑郁是该病精神症状的主要表现,一项意大利研究提示48%的CADASIL患者会出现精神症状,这些患者中出现抑郁的占69%,出现焦虑的占24%[15]。淡漠为该病患者情绪异常的另一主要表现,出现在41%的法国患者中,表现为行为动机的缺乏、对外界事物兴趣的缺乏及自主行为的减少[16]。其他少见的精神症状包括攻击行为、冲动及精神分裂样症状。这些精神症状尤其是抑郁症状是患者生活质量降低的重要因素。
可逆性脑病可出现在约10%的CADASIL患者当中,可作为患者的首发症状出现。最常见的症状是幻觉、癫痫和局灶性神经功能缺损,大部分患者的症状可在3个月内完全恢复,超过一半的患者在脑病发作前和发作中有偏头痛或偏头痛先兆的表现[17]。
CADASIL患者的眼底改变包括视网膜动脉管腔变窄、视网膜静脉管腔扩大、动静脉管壁增厚,且这些表现与颅内病变的程度相关。约10%的患者会出现癫痫发作,但较少出现癫痫持续状态,使用抗癫痫药可以有效控制症状。帕金森综合征样症状见于携带p.R1006C突变的患者,且对左旋多巴反应不佳。
上述临床表现提示患者的定位诊断在大脑半球,定性诊断提示存在脑小血管病的可能性,要验证临床的定位和定性诊断需要采取下列检查手段。
血管性检查包括血常规、血生化(血糖、糖化血红蛋白、血脂、血同型半胱氨酸水平、肝肾功能等),评估患者有无心脑血管病的危险因素。
蒙特利尔认知评估量表及简易精神状态检查量表可用作CADASIL患者整体认知功能的评估。更详细的认知功能评估包括对记忆力、执行功能、视空间、注意力、语言流畅性等认知领域的评估。汉密尔顿焦虑抑郁量表、淡漠自评量表等可用于患者精神症状的评估。改良Rankin量表评分、Barthel指数等可用于对患者日常生活自理能力和残疾程度的评估。
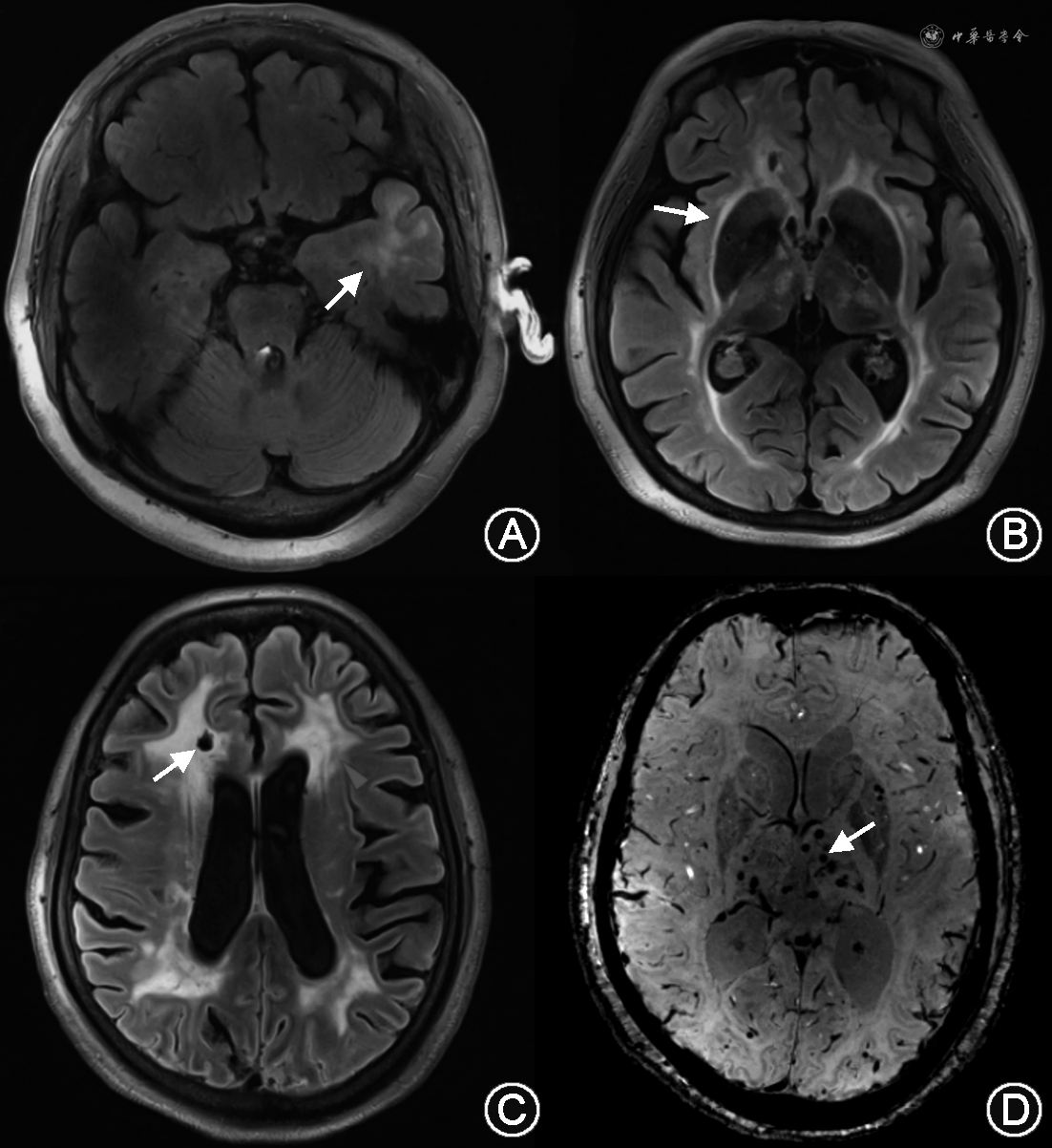
MRI检查包括常规头颅平扫序列和微出血扫描序列,其改变包括(图2):(1)脑白质高信号(white matter hyperintensities,WMHs)是患者最早和最常见的影像学改变,35岁之后基本上所有的CADASIL患者都会有不同程度的WMHs[18],分布在脑室周围白质、颞极、外囊、额顶区白质,U形纤维通常不被累及[19]。一项小样本研究结果表明,颞极白质高信号区别CADASIL和散发脑小血管病的敏感度和特异度分别为90%和100%[20],但亚洲患者颞极受累比例较欧美患者低。WMHs的发生原因与血脑屏障破坏导致的白质水肿以及白质纤维束的病变有关。(2)腔隙性脑梗死(lacunar infarctions,LIs)是患者另一常见的影像学表现,出现在几乎所有疾病晚期的患者中,主要分布于半卵圆中心、丘脑、基底节和脑桥。LIs与WMHs有一定的相关性,Duering等[21]发现大约90%的LIs出现在WMHs的边缘,仅5.8%出现在WMHs中或WMHs以外,提示LIs与WMHs是同一疾病动态发展过程中的不同表现。越来越多的研究结果提示,CADASIL患者的LIs不是由穿通动脉的闭塞造成,而可能与血流动力学改变导致的局部低灌注有关,对CADASIL患者进行7.0 T MRI检查也提示其基底节的病变可能并不是由豆纹动脉的闭塞造成[22]。(3)脑微出血(cerebral microbleeds)出现在约50%的我国患者当中,主要分布在深部脑区。脑微出血数量与患者的年龄和MRI总病灶负荷呈正相关,高血压与脑微出血的发生独立相关,脑微出血负荷(微出血数量≥9个)与脑出血的发生独立相关[23]。(4)血管周围间隙(perivascular space)的扩大可出现在78%的患者中,主要位于颞叶、基底节和皮质下白质,其严重程度随患者的年龄增长而增加,颞叶或岛叶下区扩大的血管周围间隙的严重程度与患者脑白质高信号的严重程度密切相关[24]。由于CADASIL患者多无明确的血管狭窄和闭塞,脑血管造影不推荐,个别研究报道血管造影可能会增加患者神经系统并发症的风险[25]。
对于首诊高度怀疑CADASIL的患者可进行NOTCH3基因序列分析。可以采取包括各种遗传性脑血管病基因的靶向二代测序方法,有助于疾病的鉴别诊断。对检测到的变异,应根据美国医学遗传学与基因组学学会遗传变异分类标准与指南进行致病性判断[26]。目前已报道的NOTCH3致病变异多为累及半胱氨酸的错义突变(如p.R90C或者p.C222G),所以当检测到的变异不影响半胱氨酸替换时,对其致病性的分析需慎重。
电镜下观察到皮肤小动脉血管平滑肌细胞基底膜出现GOM,可确诊为CADASIL。
CADASIL的诊断首先基于患者的临床表现和家族史,而后进行头颅MRI检查,MRI显示CADASIL影像学改变的特点时,再进一步行基因和病理检查以明确诊断。该病的鉴别诊断包括散发性和遗传性中枢神经系统疾病。
1.多发性硬化:CADASIL发病早期的症状和影像学表现与多发性硬化较为相似。多发性硬化患者脑脊液中常会出现寡克隆区带,MRI表现为白质内多发的长T2异常信号,常累及脑室旁、近皮质、胼胝体,而颞极受累少见。典型的多发性硬化脑室旁病灶呈火焰征(Dawson指),进展期病灶可强化[27]。
2.动脉硬化相关性脑小动脉病:与长期高血压、糖尿病密切相关,如Binswanger病。其临床特征是进行性加重的认知障碍、步态障碍,MRI可以发现弥漫性脑白质T2高信号、腔隙性脑梗死及微出血,与CADASIL类似,但该类疾病一般没有颞极的白质高信号[28]。
3.原发性中枢神经系统血管炎:在各个年龄段均可以发病,以中青年发病多见,常出现头痛、认知障碍和局灶性脑损害表现,头颅MRI可见皮质及皮质下白质多发性病灶,可伴随皮质、皮质下及脑膜的强化改变,通常无颞极白质高信号,脑组织病理结果是诊断该病的“金标准”[29]。
1.常染色体隐性遗传脑动脉病伴皮质下梗死及白质脑病(cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy,CARASIL):由HTRA1基因纯合突变或复合杂合突变引起。发病年龄比CADASIL略早,主要临床表现与CADASIL类似,患者常有秃头、腰痛及皮肤粗糙等神经系统外症状。头颅MRI表现为广泛脑白质病变、多发脑梗死,也可见双侧外囊和颞极的白质病变[30]。
2.HTRA1相关常染色体显性遗传白质脑病:由HTRA1基因杂合突变导致。与隐性遗传的CARASIL相比,本病发病年龄较晚,症状较轻,神经系统以外的特征也相对较少[31]。
3.伴白质脑病和全身表现的视网膜血管病(retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations):是由TREX1基因突变引起的常染色体显性遗传小血管病,发病年龄通常在35~50岁,其病变可累及神经系统以外的器官,包括眼底、肾脏、肝脏,MRI主要表现为脑室周围和深部白质的片状白质病变,病灶可伴环状或结节性强化[32]。
4.神经元核内包涵体病(neuronal intranuclear inclusion disease):多在成年晚期发病,由NOTCH2NLC基因的三核苷酸重复扩张导致,临床表现特点为头痛、意识障碍、精神障碍,可以伴随突发性失语或发热,和可逆性脑病类型的CADASIL类似。MRI显示大脑白质高信号,弥散加权成像可见沿灰白质交界区分布形成“飘带征”[33]。
CADASIL的治疗遵循脑卒中指南进行管理[34],以一级、二级预防,对症支持治疗为主,同时应注重患者的心理治疗、康复治疗和日常护理。
一级预防的主要目的在于控制脑血管病危险因素,其措施包括:适度的体育锻炼、合理膳食、防治肥胖和血脂异常、控制血压、防治糖尿病、治疗高同型半胱氨酸血症、防治心脏病、改善脑供血等。
可参照其他缺血性脑卒中的治疗方案进行治疗。急性发作期的治疗目的在于维持患者的生命体征和血压平稳,预防误吸及水电解质紊乱等并发症,改善神经功能等。缺血性事件二级预防的措施包括:积极控制心脑血管病危险因素,改善脑供血等。频繁发生缺血性事件的患者可以考虑进行抗血小板药物治疗,存在大量脑微出血和高血压脑出血的危险因素时,要慎重选择抗血小板或抗凝药物。
治疗方法与普通偏头痛类似。应注意曲普坦类和麦角类会引起脑血管收缩和血管内皮损伤,这些药物应避免应用于CADASIL的治疗。阿米替林、β受体阻滞剂、氟桂利嗪和托吡酯等用于常规偏头痛患者的预防性治疗药物也可用于该病患者,但可能会加重患者的情绪和认知症状。乙酰唑胺除可改善脑血流外,对偏头痛可能也有一定的预防作用。
常规用于治疗认知功能损伤的药物可用于治疗CADASIL的认知功能损害,但临床疗效尚不明确。多奈哌齐没有改善患者的血管性痴呆评估量表评分,但对执行功能有一定的改善作用[35]。
常规用于精神症状治疗的药物可用于CADASIL患者。抗抑郁类药物如五羟色胺再摄取抑制剂能改善患者的情绪障碍,促进患者神经功能的恢复。双相情感障碍可以尝试给予喹硫平治疗,精神分裂样症状可以用利培酮、丙戊酸钠和氟哌噻吨治疗。
症状性脑出血的处理与其他脑出血基本类似。CADASIL脑病多为自限性,临床以对症支持治疗为主,有病例报道在患者脑病急性期,使用甲泼尼龙1 g/d冲击治疗可以促进临床症状的缓解[36],但其安全性有待进一步评估。
随着CADASIL疾病的进展,患者的神经功能退化逐渐加重。有研究报道[39],男性和女性丧失独立行走能力的中位年龄分别为58.9岁和62.1岁,死亡的中位年龄分别为64.6岁和70.7岁。吸入性肺炎是CADASIL最常见的死因,其次是猝死和窒息[39]。其中突变位于第1~6 EGFr结构域的CADASIL患者,比突变位于第7~34 EGFr结构域的患者发病更早,白质病变更重,生存期更短[40]。
CADASIL为常染色体显性遗传病,先证者的兄弟姐妹及其子代的患病风险均是50%,故应仔细询问患者家族中其他成员的情况,并建议对家系中的成年个体进行基因检测。携带致病变异者如果妊娠,建议做产前诊断。
CADASIL是最常见的遗传性脑小血管病,该病的致病基因确定为NOTCH3基因,变异的NOTCH3蛋白可通过获得新的毒性功能导致血管平滑肌细胞内关键分子通路失调、细胞骨架结构紊乱、膜离子通道异常、细胞增殖能力改变,从而造成脑小动脉结构与功能异常[3, 4]。该病主要的临床表现包括反复发作的偏头痛、脑缺血事件、认知功能下降及精神症状。特征性影像学改变对该病的诊断具有提示意义。疾病的诊断需要在临床和影像学改变的基础上,结合血管病理和基因检查结果确诊。其治疗方案和其他脑小血管病类似,但需注意患者的脑出血倾向。进一步明确NOTCH3基因突变引起小动脉结构功能异常的机制,是寻找CADASIL疾病治疗靶点的关键。
所有作者均声明不存在利益冲突
None declared
1. 常染色体显性遗传头颅动脉病伴皮质下梗死及白质脑病(CADASIL)的常见头颅MRI改变不包括以下哪项?
A.多发腔隙性脑梗死
B.皮髓质交界区弥散加权成像高信号呈“飘带征”
C.脑白质高信号
D.脑微出血
2. CADASIL的致病基因是?
A. NOTCH2NLC
B. TREX1
C. NOTCH3
D. COL4A1
3. CADASIL患者皮肤活组织检查,发现以下哪种改变为病理诊断的“金标准”?
A. 小静脉周围炎性细胞浸润
B. 微小动脉内皮细胞凋亡
C. 微小动脉平滑肌细胞凋亡
D. 微小动脉平滑肌细胞表面出现嗜锇性颗粒物质
4. 对疑诊CADASIL的患者进行基因检测,发现NOTCH3基因变异。对以下变异的致病性评价,哪种变异最可能为致病性变异?
A. p.R90C
B. p.R90S
C. p.R90L
D. p.R90G
5. CADASIL的常见临床表现不包括?
A. 痴呆
B. 精神行为异常
C. 偏瘫
D. 秃头

























