
脊髓小脑共济失调2型常见临床表现有共济失调、构音障碍、扫视变慢、腱反射减弱或消失等,表现为帕金森样症状、认知障碍等的患者较为少见。帕金森样症状少见的原因可能是患者黑质纹状体系统的退变仅表现在纹状体突触前膜而突触后膜受体相对保留,同时丘脑底核和小脑的退化掩盖了帕金森样症状的出现。认知下降的原因可能与脊髓小脑共济失调2型患者小脑与大脑皮质之间的网络连接减少有关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
脊髓小脑共济失调2型(spinocerebellar ataxia 2,SCA2)是脊髓小脑共济失调(spinocerebellar ataxia,SCA)的一个亚型,是ATXN2基因的CAG异常扩增引起的常染色体显性遗传性疾病。SCA2的常见临床症状有共济失调、构音障碍、扫视变慢、腱反射减弱或消失等,帕金森样症状、认知下降等表现较为少见[1],且较易引起误诊。近期我们通过基因检测明确1例表现为帕金森样症状和认知下降的SCA2患者,现报道如下。
患者男性,61岁,因“行走缓慢17年,幻觉伴行为异常1年”于2020年11月2—12日于北京医院神经内科住院治疗。17年前患者无明显诱因逐渐出现行走缓慢,走路步伐减小,未予重视。15年前患者行走缓慢的症状逐渐加重,并出现双手震颤,遂就诊于北京某医院,诊断为帕金森病,予多巴丝肼治疗,具体剂量不详,患者症状有缓解。后患者症状逐渐进展,并多次调整用药,具体不详。近几年,患者逐渐出现入睡困难,夜间多梦,说梦话,开始出现大便干结、便秘,平均2~3 d大便1次,并有夜尿次数增多,平均每晚起夜4~5次。1年前,患者开始间断出现幻觉,自述大脑中有两个小人打架,或有人在暗中支配自己的行动等,伴有烦躁不安、随意丢弃家中物品等异常行为,曾因充错话费而摔坏手机,并有记忆力、定向力减退,外出时需要家人陪同。1个月前,患者行走缓慢的症状较前明显加重,并伴有流涎、睁眼费力等症状,无体位性头晕、失语、失用等。多巴丝肼总剂量1 687.5 mg/d,分4次服用;卡左双多巴缓释片总剂量1片/d,分2次服用;恩他卡朋总剂量400 mg/d,分4次服用;劳拉西泮每晚1 mg。用药后约半小时可起效,每次药效可维持2~3 h。起病以来患者无跌倒,无明显失语、失用,近1年体重减轻约20 kg。既往有高血压病史。有吸烟饮酒史。有家族史,母亲及弟弟患有类似疾病。
入院体检:意识清楚,言语流利,记忆力、定向力及计算力有下降。脑神经:可疑嗅觉减退,Sniffin′ Sticks嗅棒12分(满分16分),视力视野正常,双侧眼球活动充分。额纹双侧对称,右侧鼻唇沟稍浅,软腭活动正常,悬雍垂居中,咽反射正常,耸肩转颈正常,伸舌稍偏右,左利手,四肢肌力Ⅴ级,双侧上肢肌张力正常,双下肢肌张力铅管样增高,偶有双手静止性震颤。双侧指鼻及跟膝胫试验正常,Romberg征正常。双侧肢体针刺痛觉对称。皮质觉正常。双侧肱二头肌、肱三头肌、桡骨膜及膝腱反射均对称活跃,双侧Choddock征(+),双侧Babinski征(+)。无颈抵抗,Kernig征(-)。无直立性低血压。
入院后完善相关检查:简易精神状态检查(MMSE)评分21分,蒙特利尔认知评估量表评分13分(文化程度:高中),汉密尔顿抑郁量表(HAMD)评分26分,汉密尔顿焦虑量表评分20分。头颅CT示:右前额金属密度影;右枕部皮下结节。因头部金属碎片未行MRI检查。因费用问题未行脑多巴胺转运蛋白PET/CT和多巴胺受体2 PET/CT。11月6日行左旋多巴负荷试验,国际运动障碍学会-统一帕金森病评定量表第三部分(Unified Parkinson′s Disease Rating ScaleⅢ,UPDRSⅢ)基线评分24分,1 h最佳改善时评分为6分,改善率75%。多导睡眠图因患者夜间精神症状发作未完成。
患者符合帕金森综合征的诊断标准,符合2条支持标准(患者对多巴胺能药物的治疗明确且显著有效、临床体检观察到单个肢体的静止性震颤),存在1个警示征象(出现其他原因不能解释的锥体束征),诊断为临床很可能的帕金森病。患者要求行脑深部电刺激术,但因患者目前有认知功能障碍,且精神症状明显,评估后认为不适合进行脑深部电刺激术。
因患者精神症状明显,将多巴丝肼减量至 250 mg 4次/d,卡左双多巴缓释片减量至每日1次每次半片,加用劳拉西泮片 1 mg 每晚1次、富马酸喹硫平片 25 mg 3次/d。患者精神症状稍缓解,精神症状出现次数减少,但震颤较前明显。
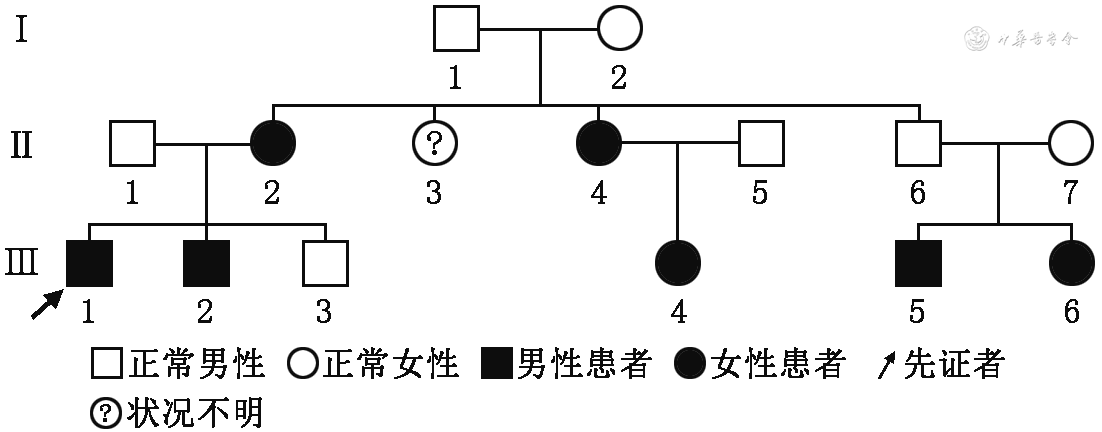
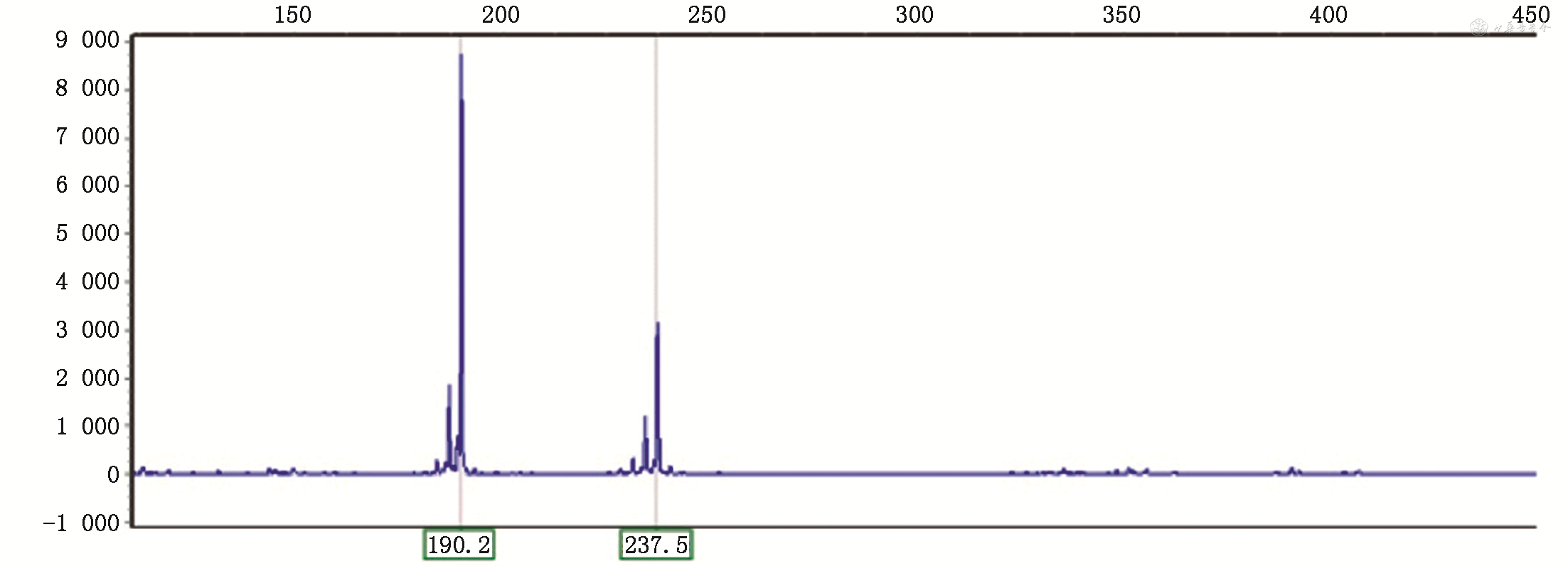
患者家族中多人有帕金森样症状,完成系谱图见图1。患者本人基因检测回报ATXN2基因2个等位基因CAG重复次数分别为20和36次(图2),符合SCA2;同时基因检测发现该患者在常染色体显性帕金森病8型相关基因LRRK2存在一处杂合变异。患者家属不同意行基因检测验证家族中其他人是否患有SCA2。
SCA2是SCA中第二大常见的类型,仅次于SCA3。SCA的全球发病率为1∶35 000,SCA2占SCA的15%[1]。SCA2是由于ATXN2基因的CAG重复序列过多,其编码的蛋白多聚谷氨酰胺扩增引起的,属于多聚谷氨酰胺疾病(polyglutamine disease)的一种。CAG重复序列≤31是正常的,其中>90%的正常人群CAG重复序列数为22次,SCA2患者的CAG重复序列数>31次。SCA2患者的发病年龄跨度较大,发病年龄早与其CAG重复序列数多有关[2]。SCA2的常见临床症状有小脑性症状(共济失调、构音障碍、辨距不良、运动性震颤)、眼部运动异常(扫视变慢、核上性眼肌麻痹)、周围神经病变(膝腱反射、踝反射减弱或消失,震动觉减退)等;病理征出现在约1/4的患者;自主神经症状也较为常见,帕金森样症状、认知下降、肌张力障碍等表现较为少见。头部磁共振成像(MRI)显示严重的橄榄体脑桥小脑萎缩(olivopontocerebellar atrophy),MRI容积测量显示小脑半球、小脑蚓部、脑桥、中脑、丘脑、额颞叶均有体积萎缩[1]。因SCA2的临床症状和影像学表现均无特异性,易与帕金森病、多系统萎缩等疾病相混淆,确诊主要依靠基因检测。SCA2患者从发病开始到坐轮椅的病程在12~25年。古巴的统计数据表明,SCA2患者从出生至死亡的时间平均为52年,从发病到死亡的时间平均为21.8年[3]。
ATXN2基因编码形成ataxin-2蛋白,早期研究发现ataxin-2蛋白仅在细胞质中表达,近期的研究发现其在细胞质和细胞核中均有表达,提示该蛋白能穿梭至细胞核以辅助调节RNA转录。ataxin-2蛋白在浦肯野细胞、脑干核团、皮质神经元等均有表达。ataxin-2蛋白在调节RNA转录及营养代谢方面均有影响。ataxin-2蛋白通过对目标RNA的影响,参与了胚胎发育、细胞凋亡、肌动蛋白发育、细胞增殖、胰岛素信号传递等多种生理过程[4]。ATXN2基因CAG重复序列过长引起ataxin-2蛋白的过度聚集,这种蛋白的过度聚集会引起神经元凋亡从而引起神经系统的退变[5]。此外,SCA2小鼠与野生小鼠相比,小脑的浦肯野细胞自发性放电的频率明显减少,表明ataxin-2蛋白严重损害了小脑的浦肯野细胞[6],而对SCA2小鼠的小脑切片发现小脑浦肯野细胞的自发性放电受损与其小电导钙激活钾通道2的异常有关[7]。SCA2患者尸检病理发现脑组织缺乏硫苷脂、半乳糖神经酰胺、神经鞘磷脂和GM1a/GD1b神经节苷脂,这些构成神经元周围髓鞘的脂质的缺乏,可能会引起神经元信号传导的异常[8]。
本例SCA2患者在发病早期主要表现为帕金森样症状,且对左旋多巴反应好。在SCA2患者中表现为帕金森样症状的较为少见。一项中国的研究纳入了135例SCA2患者,其中帕金森样症状的发生率为11.8%,80%的帕金森样症状的患者对左旋多巴有反应[9]。
对于有帕金森样症状的SCA2患者,其基因突变的特点尚不明确。有研究发现表现为帕金森样症状的SCA2患者的CAG重复序列较少,其CAG重复序列中有被CAA打断[10]。有研究者比较了分别表现为帕金森样症状和共济失调的SCA2患者,发现其CAG重复序列的长度没有明显差异,有帕金森样症状的SCA2患者CAG重复序列的长度为35~51,且仅有1例帕金森样症状患者有CAG重复序列被CAA打断的情况,这可能是因为该研究中的部分患者有帕金森样症状的同时还有共济失调[9]。一项法国的研究纳入了常染色体显性帕金森病的164个家系的178例患者,筛查SCA2、SCA3、SCA17和LRRK2基因,结果发现有3个家系9例SCA2患者,同时有10个家系18例有富含亮氨酸重复序列激酶2基因(leucine-rich repeat kinase 2 gene,LRRK2)的患者;SCA2家系的CAG序列长度为37~39,中间均被CAA打断,表现为典型的多巴胺反应性帕金森病;SCA2患者和LRRK2患者相比,双侧肢体症状更加对称,治疗后运动症状(UPDRS评分)改善较好、等效左旋多巴剂量较小且运动症状波动较小,而LRRK2患者的认知障碍更明显(MMSE评分更低);其中一对姐妹发病年龄分别为29岁和30岁,她们从母亲继承了SCA2基因同时从父亲继承了LRRK2基因杂合突变,其CAG重复序列为37次,其发病年龄较早不能用CAG重复序列长度解释,可能是因为同时伴有LRRK2基因突变使发病年龄较早[11]。一个关于表现为帕金森综合征的中国SCA2家系的研究,也发现其CAG重复序列在37~40,同时CAG重复序列中有被CAA打断,外显子测序在该家系中未发现其他突变基因,认为ATXN2基因CAG重复序列就是SCA2帕金森表型的唯一责任基因,排除了其他基因突变引起帕金森综合征表型的可能[12]。CAG重复序列较短并被CAA打断会引起SCA2患者表现为帕金森样症状的原因尚不明确,有的研究者认为:(1)这种基因特点会形成更短和更多分支的分子结构,从而影响RNA的转录和翻译;(2)这种特殊类型的基因会编码形成不同的ataxin-2蛋白,这种蛋白管理RNA翻译的功能受到了影响;(3)CAG重复序列被打断阻止了体细胞嵌合现象的发生,SCA2的体细胞嵌合现象导致小脑和脑干的细胞易受损而基底节不易受损,CAG重复序列被打断的患者其基底节多巴胺能神经元易受损,但这不能解释有不同基因特点的表现为小脑性共济失调和帕金森样症状的SCA2患者有相似的发病年龄,其分子生物学原因尚待进一步研究[11]。SCA2患者CAG重复序列数>31次,而本例患者的ATXN2基因2个等位基因CAG重复次数分别为20和36次,同时在常染色体显性帕金森病8型相关基因LRRK2存在一处杂合变异,其CAG重复序列长度符合SCA2诊断,可能是帕金森综合征表型的SCA2,而LRRK2杂合变异可能使其发病年龄较早且认知障碍较明显。
SCA2患者表现出帕金森样症状的病理生理机制尚未完全明确。一项研究纳入了6例SCA2患者、13例SCA3患者、19例原发性帕金森病患者和26名正常对照。SCA2/SCA3/帕金森病患者这3组均接受脑多巴胺转运蛋白(DAT)PET/CT和多巴胺受体2(D2受体)PET/CT显像,以比较其黑质-纹状体通路多巴胺能神经元功能。结果显示SCA2和SCA3患者与正常对照组比较其DAT显像在尾状核有明显减低,与原发性帕金森病组类似,而在D2受体显像SCA2和SCA3组没有明显减低,提示SCA2和SCA3患者的突触前DAT明显缺乏但是突触后D2受体没有明显改变。该研究还对4例SCA2患者、9例SCA3患者、13名没有神经精神疾病的志愿者共计26位已死亡的受试者进行脑部尸检,结果显示SCA2和SCA3组黑质部位多巴胺能神经元有明显丢失,与原发性帕金森病患者类似,同时SCA2和SCA3组有明显的丘脑底核神经元丢失,没有壳核和尾状核的神经元丢失。尸检的患者中仅有1例SCA3患者表现出静止性震颤的帕金森样症状并对左旋多巴反应好,其脑部尸检结果显示丘脑底核完好同时黑质被破坏[13]。根据以上研究的DAT和D2受体PET/CT及尸检发现,SCA2和SCA3患者除了小脑和脑干萎缩之外,黑质有严重退化,丘脑底核也有退化,但大部分SCA2患者却没有表现出帕金森样症状,只有1例患者生前表现出了帕金森样症状,尸检结果表明这例患者丘脑底核没有退化,提示可能是丘脑底核的退化阻止了SCA2患者帕金森症状的出现。同时SCA2和SCA3患者的PET/CT和尸检都提示严重的黑质纹状体系统的退变,但同时纹状体突触后多巴胺受体的密度没有下降,表明在SCA2和SCA3患者中这种黑质损害更加明显,与帕金森病类似,这也解释了有帕金森样症状的SCA2和SCA3的患者对左旋多巴反应良好。另外,小脑的退化可能阻止或者掩盖了帕金森样症状[14]。
虽然SCA2患者可表现出帕金森样症状,但SCA2毕竟是一种少见遗传性疾病,锥体外系疾病仍要首先考虑帕金森病、多系统萎缩等更常见疾病,并不支持在无家族史锥体外系患者中常规进行SCA2基因筛查。一项多中心大规模研究纳入12 346例帕金森病患者,发现帕金森病患者组和正常对照组在SCA2、SCA3、SCA6和SCA17基因筛查中无明显差异,不支持在原发性帕金森病患者中进行SCA基因筛查[15]。一项研究纳入270例早发或晚发多巴胺反应性帕金森样症状患者,进行基因筛查未发现有患者出现SCA2基因突变[16]。一项韩国研究包括603例有帕金森样症状的病程在3年以上患者,其中临床诊断为帕金森病的有468例,临床诊断为多系统萎缩帕金森型(MSA-P)的135例,对这些患者筛查ATXN2基因CAG重复序列,发现其中有3例患者符合SCA2诊断标准,且这3例都没有家族史和共济失调,3例中有2例临床诊断为帕金森病且对左旋多巴有明显反应,其中1例诊断为MSA-P且对左旋多巴治疗反应很小[10]。
本例患者除在发病早期表现出帕金森样症状之外,在发病晚期出现了明显的认知下降。认知下降在SCA2患者中并不常见。一项中国研究纳入了135例SCA2患者,其中认知下降的发生率为14.7%[9]。作为少见的SCA2症状类型,认知下降患者的基因特点尚不明确。一项印度研究纳入了73例SCA2患者,发现伴有认知障碍的患者CAG重复序列较长[2]。另一项研究纳入了48例SCA2患者,其中12例有认知障碍且全部有线粒体基因A10398G突变,比较发现有认知障碍的SCA2患者组和无认知障碍的SCA2患者组之间发病年龄、CAG重复序列长度无明显差异,但有认知障碍的SCA2患者组线粒体基因A10398G突变比例明显高于无认知障碍的SCA2患者组,这种线粒体基因的多态性可能会影响线粒体基质pH值和细胞内钙流动,也可能影响细胞核DNA的酸碱反应或内环境因子,另一方面ATXN2基因编码形成的ataxin-2蛋白与细胞器的交互作用可能选择性上调了某类线粒体,最终影响了SCA2的认知表型[17]。4例SCA2患者尸检发现所有患者均有基底前脑胆碱能神经元的大量缺失,基底前脑的神经纤维与中脑、间脑、边缘系统和大脑皮质存在广泛联系,基底前脑胆碱能神经元与记忆、学习、视空间等认知功能和睡眠周期调节等有关,这种神经元的凋亡可能与认知功能下降有关[18]。有研究结果显示SCA2患者在执行功能、语言记忆、视空间[19]、抑郁淡漠[20]等方面与正常人群比较有明显缺陷,同时SCA2患者认知障碍与运动症状之间无明显相关,提示SCA2患者认知和运动的损害可能是不同的退变机制引起的[20]。
SCA2患者表现出认知下降的机制是研究中比较热点的问题。1998年Schmahmann和Sherman[21]首次提出小脑认知情感综合征(cerebellar cognitive affective syndrome)的概念,表现为:(1)执行功能的缺陷,包括规划、变换、抽象推理、工作记忆以及语言流利性的下降;(2)空间认知受损,包括视空间混乱和视空间记忆受损;(3)人格改变,包括淡漠、脱抑制或不当行为;(4)语言障碍,包括音律失调、语法缺失、轻度的命名障碍。小脑引起的认知下降主要与小脑后叶和小脑蚓部相关,小脑前叶主要与运动相关。小脑引起认知下降可能与小脑与大脑之间的联系环路受损有关,小脑与大脑的前额叶、顶叶后部、优势半球颞叶、边缘叶相关[21]。小脑和大脑皮质之间主要的神经环路:传入通路从大脑皮质的神经突触发出,经过同侧的脑桥交叉至对侧的小脑皮质;传出通路从小脑齿状核的第一级神经突触发出,交叉到对侧的丘脑并发出第二级神经突触连接到大脑皮质[22]。一项研究纳入9例SCA2患者,所有患者均表现为典型的小脑症状,对照组包括33名健康受试者,进行静息态功能MRI和神经心理评估,两组比较发现SCA2患者的小脑后叶与大脑皮质认知心理相关的额上回和额中回部位的连接有减少;同时发现小脑前叶掌管运动的区域与大脑皮质掌管运动的区域如中央前回、中央后回以及辅助运动区域的连接也有减少,提示SCA2患者的运动和认知症状可能与小脑与大脑皮质之间的网络连接减少相关[23]。其他关于SCA2患者静息态功能MRI的研究也支持小脑皮质和大脑皮质额顶叶之间联系的减少与认知下降有关[24]。有研究者比较SCA2患者与健康对照的头颅MRI显示的小脑白质萎缩和神经心理量表,发现SCA2患者小脑后叶的白质萎缩与视空间、非文字记忆和执行功能相关,提示SCA2患者认知障碍可能与该疾病导致的小脑皮质退行性改变有关,且头部MRI证明不同区域小脑皮质改变与特定的认知症状有关,即与小脑-大脑皮质神经环路异常有关[25]。
本例患者在发病早期以帕金森样症状为主,左旋多巴治疗有效,后出现锥体束征、认知障碍、精神症状等,临床上易与帕金森病、多系统萎缩、路易体痴呆等疾病相混淆。SCA2的常见临床症状有共济失调和构音障碍等小脑症状、眼部运动异常、周围神经病变等,病理征出现在约1/4的患者;以帕金森样症状起病的SCA2患者多发病年龄较早,有明确家族史,对左旋多巴反应良好[26]。该患者44岁发病,主要表现为帕金森样症状和认知下降,无共济失调等小脑症状,有病理征,有明确家族史,左旋多巴长期治疗有效,经基因筛查明确SCA2诊断。
综上所述,本例患者所表现出的帕金森样症状和认知下降在SCA2患者中较为少见,帕金森样症状少见的原因可能是SCA2患者黑质纹状体系统的退变仅表现在纹状体突触前膜而突触后膜受体相对保留,同时丘脑底核和小脑的退化掩盖了帕金森样症状的出现;认知下降的原因可能与SCA2患者小脑与大脑皮质之间的网络连接减少有关。
所有作者均声明不存在利益冲突
None declared

























