
总结1例6q25.3缺失致Coffin-Siris综合征1型患儿的临床资料及基因突变特点。患儿为7岁6个月女童,有喂养困难、反复感染、语言及运动发育迟缓、智力低下、喉软骨发育不良等表现,浓眉、牙齿稀疏、背部多毛,伴多动及攻击性行为、癫痫发作、共济失调。先证者染色体核型分析未见异常;基因组拷贝数变异测序(CNV-seq)示染色体6q25.3区域存在约4.27 Mb杂合缺失,包含ARID1B基因在内的17个基因,其父母CNV-seq无异常。家系全外显子基因测序示先证者ARID1B基因第1~20外显子全部缺失,父母为野生型。先证者临床症状较重,单倍剂量不足是先证者的遗传学病因。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Coffin-Siris综合征(Coffin-Siris syndrome,CSS)是一种常染色体显性遗传的罕见性疾病[人类孟德尔遗传数据库(OMIM):305100],于1970年由Coffin和Siris[1]首次报道,包括CSS 1~11共11种亚型,分别由ARID1B、ARID1A、SMARCB1、SMARCA4、SMARCE1、ARID2、DPF2、SMARCC2、SOX11、SOX4、SMARCD1基因缺陷引起。目前全球报道有200余例,男女患病率无明显差异[2, 3, 4]。研究发现SMARCA2、PHF6基因的致病性变异也可能与CSS相关[5]。CSS的主要临床特征为全面发育迟缓、特殊面容、喂养困难、行为异常、发育畸形、第五指/趾发育不全、听力障碍、癫痫发作及感染等症状[6, 7]。
CSS1亚型临床罕见,目前报道较少。2012年Santen等[3]报道ARID1B基因突变可导致CSS 1亚型,约60%以上为ARID1B基因点突变,少数患者存在大片段缺失。ARID1B基因在多种细胞中表达,在大脑发育过程中发挥重要作用[5,8]。我们报道1例因6q25.3缺失导致ARID1B基因全部外显子杂合缺失、染色体片段缺失导致ARID1B基因单倍剂量不足引起的CSS 1型患者的临床资料,为该疾病的临床诊断和遗传学咨询提供依据。
临床资料 先证者为7岁6个月女童,因“全面性发育落后7年余,抽搐发作3年”于2019年6月就诊于郑州大学附属儿童医院神经内科。患儿2月龄发现追听、追视差,3个月不能竖头,4个月不会翻身,7个月不能独坐;按“发育迟缓、脑瘫”康复训练及营养脑神经治疗1年,语言及运动发育仍落后,1岁9个月会走,2岁只会喊“妈妈”,后逐渐会说3~4个词语,目前只能间断说出7~8句长字句。2岁5个月时有佝偻病样表现,脊柱弯曲。期间患儿因反复“重症肺炎、酸中毒、呼吸衰竭”在新生儿重症监护病房多次住院治疗。3岁时高热惊厥1次;4岁时出现癫痫发作,表现为全面强直-阵挛发作,2~3个月发作1次,每次持续3~5 min,给予“左乙拉西坦片、丙戊酸钠口服液”治疗,抽搐控制不佳。个人及生长发育史:系孕1产1,足月剖宫产,出生体重2.8 kg,围生期无缺氧窒息病史,母孕早期有保胎史。出生后有先天性喉软骨发育不良、喂养困难、体重未见增加、落后于正常同龄儿,对牛奶过敏、乳糖不耐受,给予无乳糖配方奶粉喂养2年,效果欠佳。家族遗传史:父亲体健,非近期结婚。体格检查:头围50 cm,体重17 kg,身高110 cm,意识清楚,身材消瘦,皮肤无咖啡斑及色素脱失斑;背部毛发较多,前额发际较多,眉毛浓密、上抬,前额突出、鼻梁扁平,上唇薄、下唇厚,牙齿稀疏、乳牙未脱落,小下颌。神经系统检查:言语少,不能完全对答,理解力及计算力困难。双侧眼睑无下垂、眼球活动正常、无眼震,颈软、无抵抗,脑神经检查无异常;四肢肌肉无萎缩,四肢肌力Ⅳ级,肌张力低下,闭目难立征(+),指鼻试验(+),跟膝胫试验、快速轮替试验(+),双侧巴宾斯基征、奥本海姆征、戈登征(-),双侧膝腱反射、踝阵挛(-)。实验室及相关辅助检查:血生化、血氨、乳酸、铜蓝蛋白、甲状腺功能均无异常;血、尿氨基酸遗传代谢筛查及心脏、腹部超声均未见异常。患儿7岁时韦氏智力测试智商数为60分(正常值>90分),中度低下。患儿头颅MRI、MRA、MRV检查结果均未见异常。长程视频脑电图示:清醒期广泛性2~3 Hz棘波、棘慢波发放。
在患儿家属签署知情同意书后,采集患儿及其父母外周静脉血5 ml,提取基因组DNA进行外周血染色体核型分析、家系全外显子基因组测序(trio-WES)、基因组拷贝数变异测序(CNV-seq),对分析得到的可疑致病变异进行Sanger测序验证。结果显示先证者外周血染色体核型分析未见异常,基因组拷贝数变异(CNV)检查结果显示6q25.3(chr6∶155642819-159912910)位置存在约4.27 Mb杂合缺失,该CNV在正常人类基因组结构变异数据库(DGV)中未见报道,变异区域涵盖基因包含ARID1B、EZR、RSPH3、GTF2H5、SERAC1、C6orf99、DYNLT1、FNDC1、NOX3、SNX9、SYNJ2、SYTL3、TAGAP、TMEM181、TMEM242、TULP4、ZDHHC14等相关基因,而ARID1B基因为单倍剂量敏感基因,关联CSS1型(OMIM:135900),基因库危害等级提示可能致病(图1A)。其中TMEM242、ZDHHC14基因与本病致病性无相关性,无ClinGen库注释。先证者父亲、母亲CNV-seq均正常,为野生型(图1B、C)。trio-WES显示患儿经荧光定量PCR验证在该区域为杂合缺失,ARID1B基因存在第1~20外显子缺失(NM_001374820),父母基因型均为野生型(图2)。该缺失变异为功能丧失性变异(超强致病证据PVS1)及新发变异(强致病证据PS2),在突变数据库、千人基因组及外显子组整合数据库等正常人群数据库中均未见收录(中等致病证据PM2)。根据2015年《美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南》,该变异分级被综合评定为致病变异[9]。
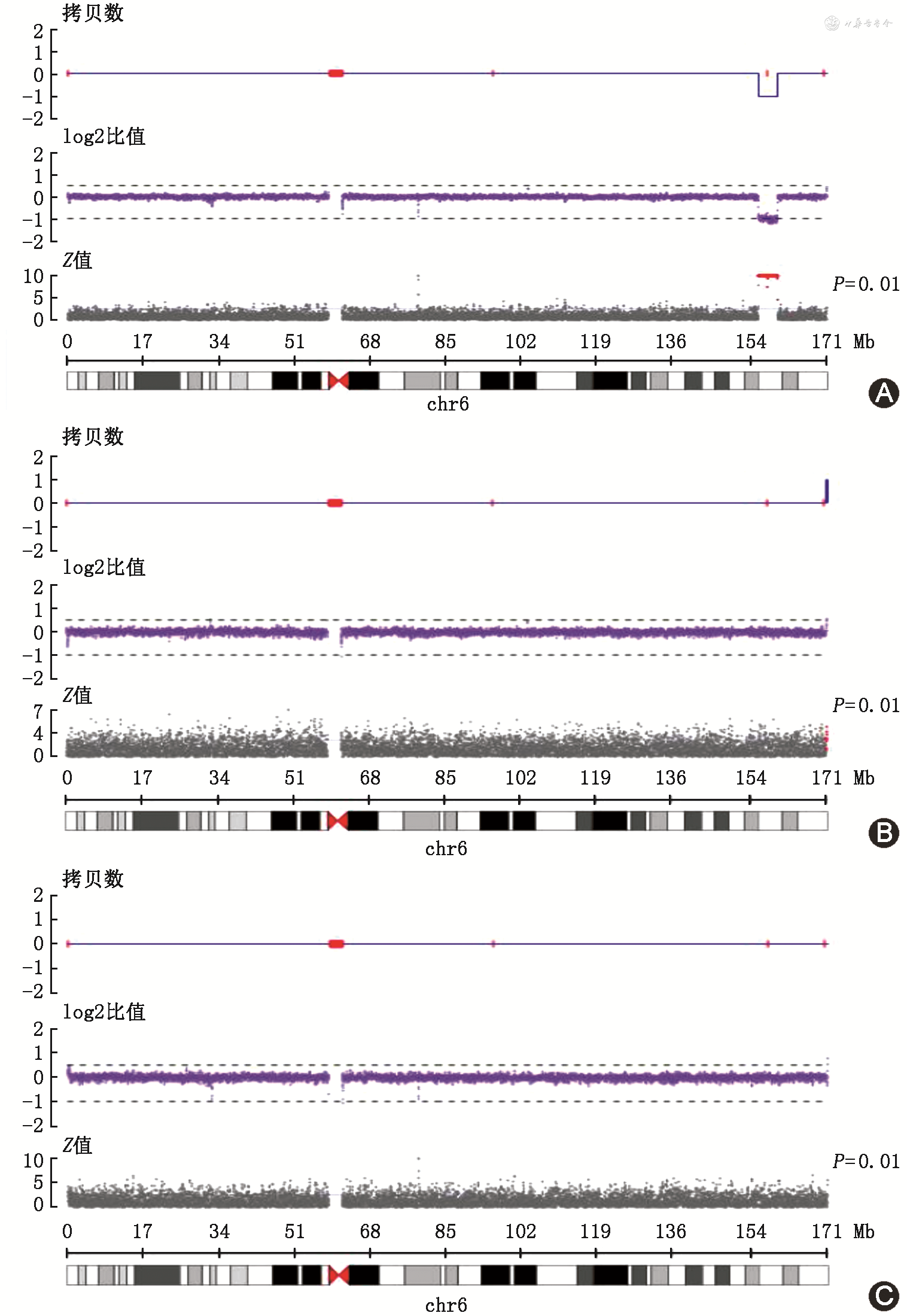
纵坐标0、1、2、-1、-2分别表示拷贝数正常、单倍重复、双倍重复、杂合缺失、纯合缺失;log2比值:检测量/对照量的log2值,表示和对照相比的差异程度;Z值:经过Z检验后,计算得到的检验值和对照组的差异,该值越大,表示和正常对照的差异越显著
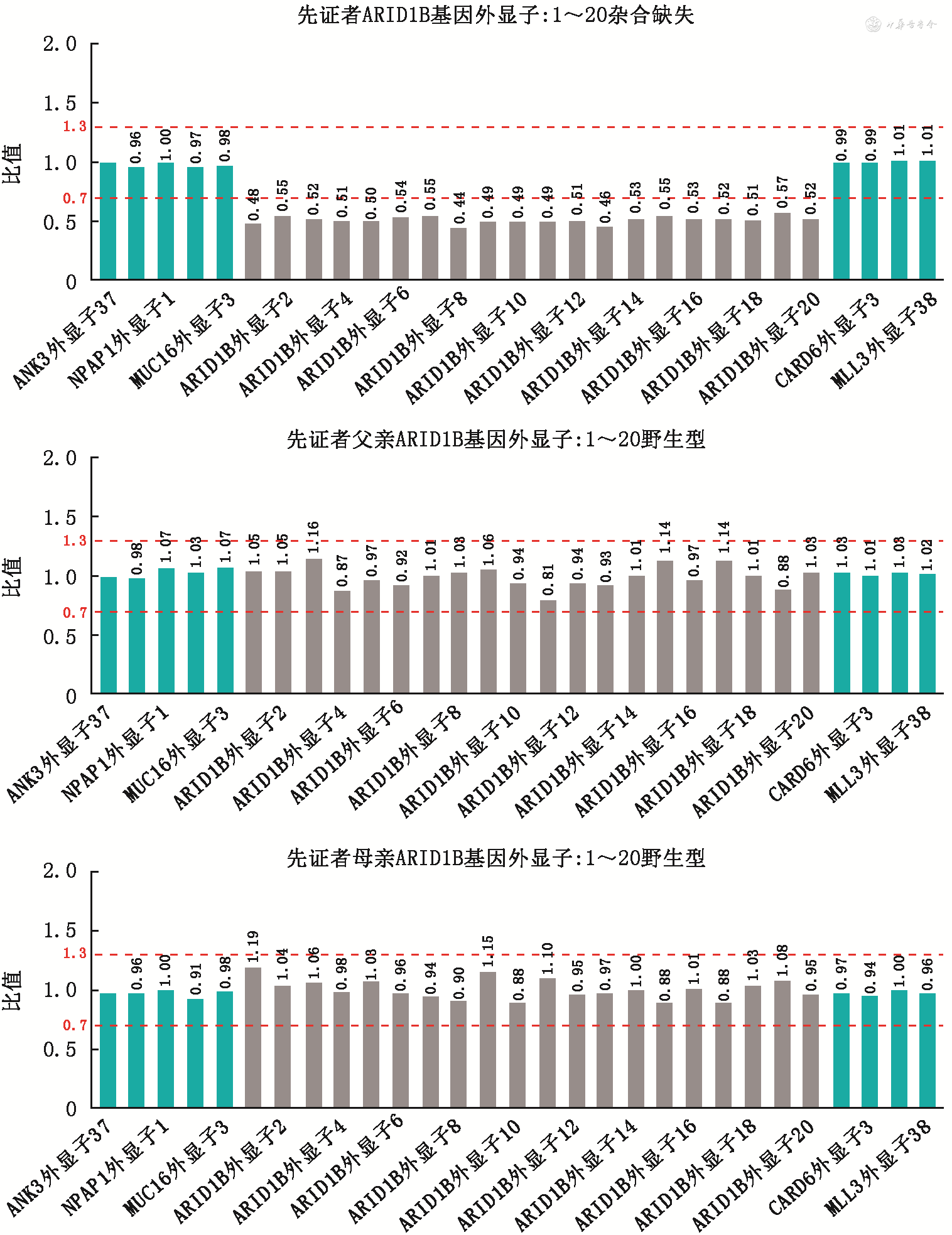
比值:检测值与对照的比值,即受检者外显子检测信号量/对照样本外显子检测信号量
根据患儿临床表现及基因测序结果,“CSS1型合并癫痫发作”诊断明确,停用丙戊酸钠口服液治疗,继续予左乙拉西坦片治疗逐渐加量至40 mg·kg-1·d-1 2次/d,控制不佳,加用拉莫三嗪至3 mg·kg-1·d-1治疗后,现患儿无癫痫发作6个月余,随访患儿语言及运动功能较前稍有进步,对颜色感兴趣,仍伴有协调障碍、步态不稳,无法单腿跳,学习困难、注意力不集中,伴有多动、自闭及攻击性行为。
CSS是一种罕见的常染色体显性遗传综合征[1, 2],主要表现有全面性发育迟缓(100%;智力或认知发育障碍、语言发育迟缓)、特殊面容(95%;鼻梁宽阔、眉毛粗、头发稀疏、嘴宽)、多毛症(95%)、身材矮小(90%)、喂养困难(90%)、远端指骨或第五指/趾甲发育不全(73%)、低钾血症(75%)、先天性心脏病(35%)、泌尿生殖系统畸形(35%)、神经系统发育异常(肌张力低下75%、癫痫发作50%、听力障碍45%、视力障碍40%、胼胝体发育不良35%)、行为异常(60%;多动、自闭及攻击性行为、学习困难、注意力不集中)[10, 11, 12]。
有研究发现CSS的发生主要与AIRD1A、AIRD1B、SMARCA4、SMARCB1、SMATCE1、SOX11基因相关[3, 4,13],其中ARID1B基因是目前CSS1型以及非特异性智力障碍患者主要的致病基因[14, 15]。酵母交配型转换/蔗糖不发酵复合物为表观遗传调控酶,在细胞中调控大量的基因表达可导致CSS的发生[16]。
ARID1B基因定位于染色体的6q25.3 区,富含AT结构域蛋白1B,全长约433 kb,有2 249个氨基酸,包含20个外显子,在活性调控及多种细胞表达中起重要作用[17]。ARID1B基因的2个结构域均参与哺乳动物大脑内的神经元分化和成熟过程[18],在大脑发育早期发挥重要作用,可使树突分支及末梢附着神经元下降[19]。在ARID1B基因变异的小鼠模型中,杂合子小鼠的γ-氨基丁酸能抑制性突触传递受损,导致中间神经元数量减少,进而导致患儿出现癫痫发作、智力发育障碍[20, 21]。
本例患儿具有较严重的语言及运动迟缓、智力发育障碍及特殊面容表现,有癫痫发作、反复感染、行为发育异常等临床表型,结合患儿的CNV-seq、trio-WES测序结果及其临床表型,诊断为CSS1型明确。通过家系CNV-seq发现,患儿6q25.3区段(chr6∶155642819-159912910)存在约4.27 Mb大片段杂合缺失,先证者父母CNV-seq均正常,为野生型,符合家系遗传共分离特点。该CNV在DGV普通人数据中未见报道,关联Decipher、ClinVar、OMIM、ClinGen、ISCA等相关数据,依据2019年的ACMG CNV解读与报告技术标准诊断指南综合评定CNV为致病性变异,查阅国内外文献及相关数据库,少数CSS患者存在6q25.3区段大片段杂合缺失[6,8],国内尚未见相同区段的外显子缺失的病例报道。
ARID1B基因的致病位点绝大多数为点突变,少数有剪切、移码、无义突变及全部基因或多个外显子缺失导致单倍剂量不足的功能缺失突变[3,12,22]。错义突变少见,可能与大多数移码变异和无义变异会激活无义介导的mRNA降解有关[19]。ARID1B基因CNV与点突变类似,会出现相应的临床表现,其可能原因是缺失1个拷贝数会引起ARID1B基因单倍剂量不足,导致先证者出现异常的临床表型[7, 8]。国外报道的CSS多由ARID1A、ARID1B、MARCA2等基因点突变导致,国内则对CSS的报道较少[6, 7],而染色体片段缺失引起ARID1B基因单倍剂量不足而导致CSS 1型的报道则更为罕见[8,18]。
本例患儿的染色体6q25.3区段存在1个拷贝缺失,该区域包括ARID1B、TMEM242、ZDHHC14等基因,该缺失导致ARID1B基因1~20外显子全部杂合缺失[氨基酸改变4.29 Mb(NM_001374820)],缺失区域经过荧光定量PCR方法验证为新生突变(de novo),父母基因型均为野生型。ARID1B在OMIM数据库中的关联疾病为CSS(OMIM:305100),数据库中收录的CSS患者表型与本例患儿高度吻合,为常染色体显性遗传。因此,考虑ARID1B基因1~20号外显子缺失引起单倍剂量不足是导致患儿异常临床表型的原因。
CSS患儿的临床表型存在异质性,不同年龄段的临床表型并不一致。本例患儿伴有癫痫发作,结合其头颅MRI、视频脑电图表现,诊断为特发性癫痫、全面性发作明确,全外显子组测序并未发现患儿存在与癫痫相关的基因变异。复习文献提示,既往有50%ARID1B基因变异致CSS的患者存在癫痫发作的相关报道[9]。因此,癫痫发作的原因以及发作类型与ARID1B基因突变可能有关。本例患儿的语言及运动功能较前稍有好转,考虑与给予抗癫痫药物治疗后癫痫发作得到控制及正规的康复治疗有关。
Mignot等[23]报道了1例母子均为轻度智力障碍伴胼胝体异常的家系,携带ARID1B基因c6092T>C(p.Ile2031Thr)杂合变异,提示该变异可能是胼胝体异常合并智力发育障碍的重要遗传因素。本例患儿头颅MRI未见胼胝体发育不全或缺如,心脏彩超及泌尿系统彩超未见先天性心脏病及泌尿生殖系统畸形,无小头畸形、视力发育异常等,与上述报道不完全一致[23]。
CSS患儿具有智力低下表现,多为中到重度,智商一般在40~69,仅有少数CSS患儿智商达到90[3]。本例患儿智力发育(智商60分)为中度低下,语言发育落后于同龄儿,且出现自闭、多动、攻击性行为,与相关文献报道基本一致[3,12]。患儿幼时存在反复感染病史,多次肺炎合并呼吸衰竭,目前患儿运动发育较前有所好转,无生长发育迟缓(矮小)的相关表现,但不排除因患儿年龄较小的原因,尚需要进一步随访。同时患儿有佝偻病表现,而CSS1型是否合并佝偻病症状有待积累更多病例进一步观察。
对于CSS患者,目前临床尚无特殊有效的治疗方案[24],主要是对症及相关支持治疗,治疗目标是提高患儿幼年的生活质量。本病在确诊后需先进行多学科会诊评估,并定期随访监测。建议患儿父母每3~6个月定期随访评估,并持续对患儿进行康复功能训练。
综上所述,本例患儿经家系CNV-seq发现ARID1B基因在6q25.3区段存在约4.27 Mb的杂合片段缺失,6q25.3区域缺失1个拷贝,缺失区域经验证为新生突变,该CNV大片段缺失导致ARID1B基因1~20号外显子杂合缺失,考虑ARID1B基因单倍剂量不足是CSS1型的主要发病原因。因此,对于该类患儿应尽早行基因诊断,从而为后续治疗及遗传咨询提供帮助,更好地判断患儿预后。
北京智因东方转化医学研究中心有限公司的协助
所有作者声明不存在利益冲突
None declared

























