Christianson综合征是一种由SLC9A6基因突变的罕见的X连锁疾病。临床表现为男性发育迟缓、语言障碍、癫痫发作、智力障碍、共济失调、小头畸形等。现报道2例男性Christianson综合征患儿:先证者1岁11个月,临床表现为小头畸形、全面发育迟缓及癫痫发作,脑电图提示中央中线区棘慢波发放,全外显子测序检测到SLC9A6基因chrX:135084373处出现突变[c.803+1(IVS6)G>A];先证者之兄4岁8个月,临床表现类似,脑电图提示双侧Rolandic区棘波、棘慢波、多棘慢波发放,磁共振成像提示存在脑萎缩,基因验证结果与先证者一致。SLC9A6基因c.803+1(IVS6)G>A剪切突变为该家系致病突变。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
Christianson综合征(Christianson syndrome,OMIM300243)是一种罕见的X连锁疾病,最早是由Christianson等[1]在1999年首次报道,其临床特征包括男性发育迟缓、语言障碍、癫痫发作、智力障碍、共济失调、小头畸形等,影像学提示可存在小脑萎缩,其表型的严重程度与分子遗传特征有关,而基因检测发现SLC9A6基因变异具有重要诊断价值,现总结1个家系中2个同胞兄弟携带SLC9A6(编码NHE6蛋白)基因杂合突变的Christianson综合征的临床表现及基因筛查结果,并结合既往文献进行分析,旨在提高临床医师对此病的认识。
临床资料 例1为先证者,男性,1岁11个月,因“反复抽搐发作1年”于2020年4月在安徽省儿童医院神经内科住院。患儿为其母亲第2胎第2产,出生胎龄37周,顺产,出生体重3 900 g,出生史无特殊。6月龄开始抬头,现仍不能独坐,翻身,与人无眼神交流,不主动抓物,不能言语。患儿自幼喂养困难,张口流口水,咀嚼能力差。癫痫表现为全面性发作,院外予以口服左乙拉西坦口服液(500 mg,每日2次)抗癫痫治疗,目前每月均有发作1~2次。入院体格检查:头围43 cm(正常均值-3倍标准差),体重9 kg(正常均值-2倍标准差),身高80 cm(正常均值-2倍标准差),意识清楚,精神反应一般,表情欠灵活(图1A),全身未见色素脱失斑,毛发颜色正常,心肺腹体格检查未见异常,四肢肌力Ⅴ级,肌张力偏低,双侧肱二、三头肌腱反射及膝腱反射可引出,病理征阴性。辅助检查:血常规、生化、心肌酶谱、血氨、丙酮酸均正常。血液联质谱及尿有机酸分析未见异常。脑脊液常规、生化未见异常。视频脑电图提示中央中线区棘慢波发放(图2A)。头颅磁共振成像(magnetic resonance imaging,MRI)未见明显异常(图1B、C)。Griffiths发育量表评估提示全面发育落后,各领域均低于同龄儿第1百分位水平,听力-语言领域发育年龄为1月龄,运动领域发育年龄为5月龄。
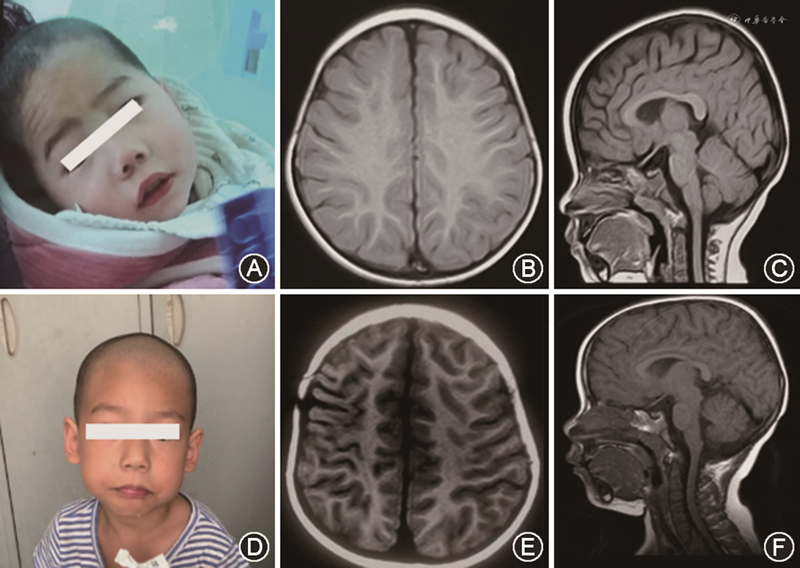
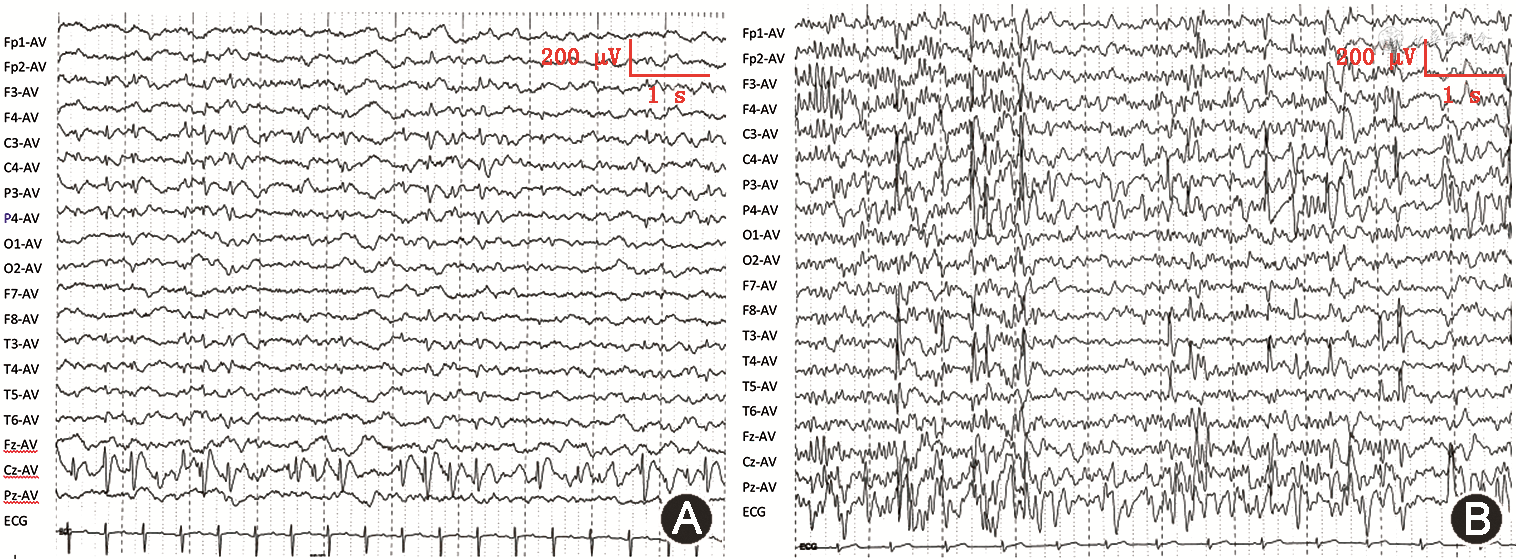
例2为先证者之兄,男性,4岁8个月,足月顺产,生后出现全面发育迟缓,8月龄独坐,18月龄可扶行,现走路容易跌倒,与人眼神交流差,不能有效沟通,不能认识家人,院外曾考虑诊断“自闭症”,予以康复锻炼,进步缓慢,40月龄时开始出现抽搐发作,均在睡眠中出现,表现为呼之不应,双目上视,牙关紧闭,口吐白沫,口唇发绀,四肢强直抖动,持续1~3 min自行缓解,在当地医院诊断为“癫痫”,予以口服丙戊酸钠口服液(280 mg,每日2次)抗癫痫治疗,服药后未再出现抽搐发作。体格检查:头围46 cm(正常均值-3倍标准差),体重13 kg(正常均值-2倍标准差),身高96 cm(正常均值-2倍标准差),表情欠灵活(图1D),心肺腹体检未见异常,四肢肌力Ⅴ级,肌张力偏低,膝腱反射可引出,病理征阴性。辅助检查:视频脑电图示双侧Rolandic区棘波、棘慢波、多棘慢波发放(图2B)。头颅MRI平扫提示存在萎缩性改变(图1E、F)。Griffiths发育量表评估提示全面发育落后,各领域均低于同龄儿第1百分位水平,听力-语言领域发育年龄为8月龄,而运动领域发育年龄为12月龄。
患儿父母均身体健康,非近亲结婚,无类似疾病家族史,患儿母亲28岁,高中文化水平,体格检查:头围54 cm,体重123 kg,身高165 cm,表情正常,对答切题,心肺腹体检未见异常,四肢肌力肌张力正常,膝腱反射可正常引出,病理征阴性。
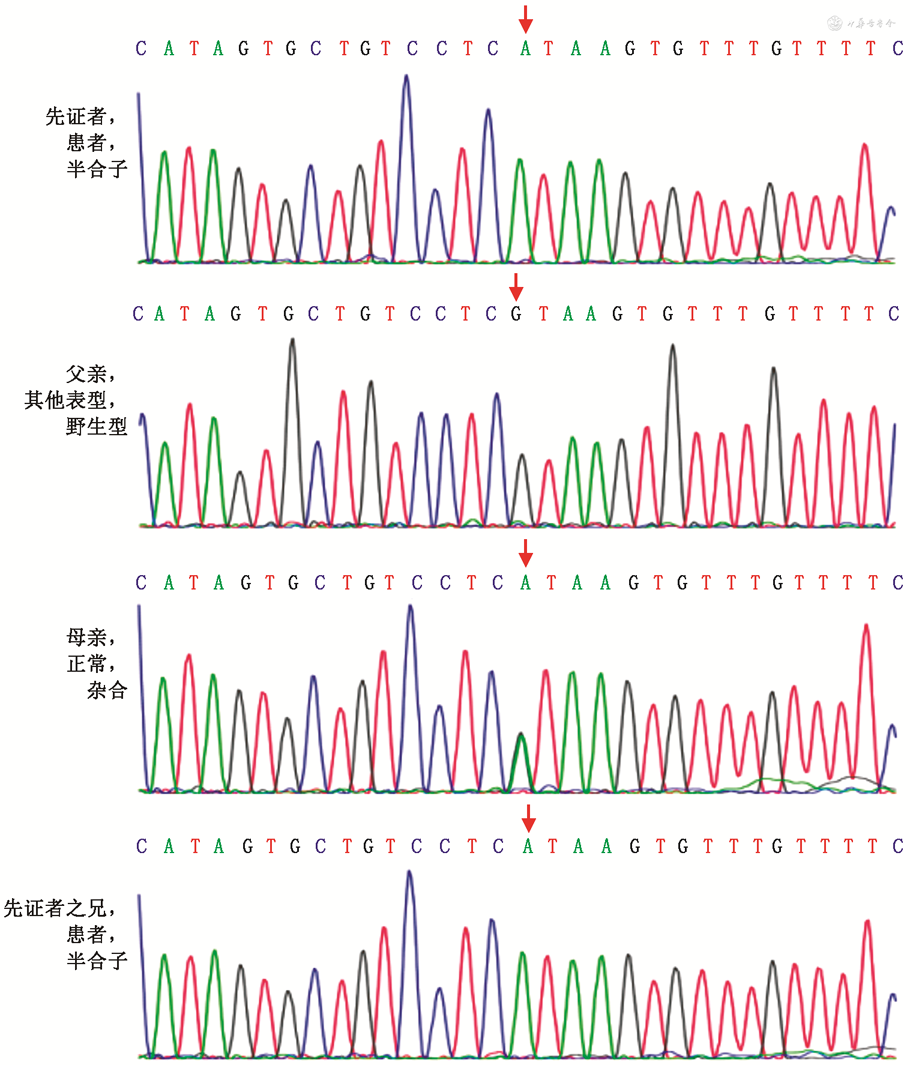
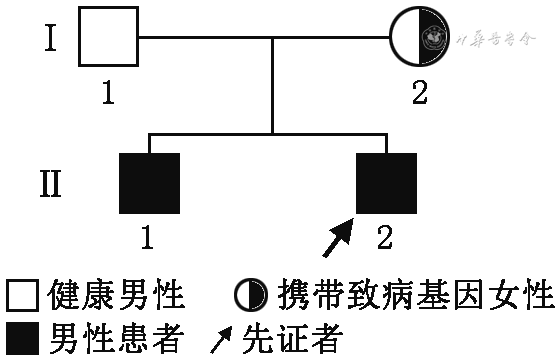
经安徽省儿童医院伦理委员会审核批准(批号:EYLL-2014-01)和患儿父母签署知情同意书后(基因检测委托北京智因东方检验实验室),采集先证者外周静脉血进行二代测序,结果提示其SLC9A6基因chrX:135084373处出现c.803+1(IVS6)G>A(NM_006359),为剪接突变。在知情同意的情况下,抽取先证者兄长及父母外周静脉血,经Sanger测序进行验证及来源分析,结果提示其兄长具有同一变异位点,母亲为该变异携带者,父亲为野生型(图3),家系图见图4。该变异先证者为半合子,符合X染色体显性遗传疾病发病机制,根据美国医学遗传学与基因组学学会(American College of Medical Genetics)变异解读指南,该变异为致病变异。
治疗经过及随访:由于先证者口服左乙拉西坦(500 mg,每日2次)治疗效果不佳,在门诊复诊过程中加用丙戊酸钠口服液(160 mg,每日2次)联合抗癫痫治疗,通过电话随访发现患儿服药2个月后未诉再有抽搐发作。先证者之兄在随访的半年内,未再出现癫痫发作,药物治疗后控制较好,但复查脑电图仍以Rolandic区放电为主,非快速动眼期棘慢波指数(spike-wave index,SWI)增加,院外继续康复治疗并接受特殊教育学校学习,认知能力较前略有改善,愿意与小朋友主动玩耍,能听懂大人部分指令。
Christianson等[1]于1999年首次描述了1个X连锁智力障碍的五代南非家庭中存在的多个受影响的男性患者及女性携带者,男性患者有着严重的智力低下、癫痫、颅面畸形、小头畸形、双侧眼肌麻痹、共济失调等临床特点,而其中一些女性携带者患有轻度智力障碍,后被命名为Christianson综合征。至2008年,更多的研究者发现该综合征与Angelman综合征(OMIM105830)有许多临床特征重叠,包括发育迟缓、小头畸形和异常的快乐行为,从而导致疾病早期容易被诊断为Angelman综合征,临床鉴别较为困难,但基因检测是其确诊的重要依据,并提出SLC9A6基因为候选致病基因[2, 3, 4]。Garbern等[5]于2020年通过神经病理学研究发现,SLC9A6基因的半合子突变是导致Christianson综合征的致病基因。目前经查询数据库及相关文献,至少已鉴定出35种不同的SLC9A6致病变异,其中包括16个外显子变异和10个内含子剪接位点变异[6]。本文报道的2例患儿SLC9A6基因存在中相同的剪接位点变异c.803+1(IVS6)G>A,临床上表现出与文献报道相似的临床特征,如小头畸形、全面发育迟缓及癫痫发作。
Pescosolido等[7]于2015年根据对12个家庭的一项研究进行分析后,提出了Christianson综合征的核心及次要诊断标准(这些临床特征存在于85%以上的受影响男性患者):核心临床表现(>85%的患者出现)包括语言障碍、智力低下(中度至重度范围)、癫痫、共济失调、产后小头畸形和(或)头围生长缓慢、多动行为;次要临床表现(>35%的患者出现)包括自闭症、类似Angelman综合征表现(尤其在出现症状前5年)、眼球活动障碍(如斜视)、肌张力减退(特别是10岁后丧失行走能力)、低身高/体重、MRI提示小脑萎缩、胃食管反流。与Angelman综合征相比,Christianson综合征的显著特征是眼肌麻痹或眼斜视、自闭症样行为、进行性小脑萎缩和精神运动发育迟缓,这些症状都会发生在50%以上的患者,尤其在出现临床表现的第一个10年后[8]。另外,在脑电图上Angelman综合征的典型脑电图特征为醒睡各期前头部、后头部及广泛性δ及θ节律性阵发或连续发放,慢波在多部位之间常呈游走性[9],但不是每个Angelman综合征患者都会出现癫痫发作,而在Christianson综合征患者中癫痫发作更为常见。
癫痫是Christianson综合征的一个显著的特征,而且大部分患者对抗癫痫药物耐药,通常在3岁之前开始出现发作,可包括婴儿痉挛、强直阵挛、肌阵挛和强直性发作等多种发作形式,发作间期脑电图癫痫样放电可为局灶性、多灶性或广泛性。Zanni等[10]于2014年报道SLC9A6基因突变可导致Christianson综合征患者出现睡眠期癫痫性电持续状态(electrical status epilepticus during slow-wave sleep,ESES)。本次报道的先证者之兄于40月龄时出现睡眠中癫痫发作,同时脑电图提示Rolandic区异常放电,复查时非快速眼球运动期SWI增加,出现ESES趋势。GRIN2A基因被认为是与ESES关联性最强的基因,位于16p13.2染色体上,编码N-甲基-D-天冬氨酸受体的GLuN2A亚单位,N-甲基-D-天冬氨酸受体不仅在突触兴奋性传递、突触可塑性和大脑发育中起关键作用,在癫痫的发病中也有越来越重要的影响[11]。ESES中报道的许多临床特征已经在Christianson综合征患者中被描述,包括认知障碍、自闭症特征。总结文献中的病例,发现在同一神经发育年龄窗口中,GRIN2A和SLC9A6基因相关疾病可能有着共同紊乱的谷氨酸信号通路[12],所以说SLC9A6基因突变可能是导致ESES的新原因。
在Christianson综合征患者中,小脑蚓部、小脑半球和海马的萎缩是最常见的MRI表现,在磁共振波谱上可发现基底节区谷氨酸/谷氨酰胺升高,可同时伴随着神经发育倒退[13]。而神经病理学研究结果表明,小脑萎缩是由浦肯野细胞的神经元丢失引起的,在SLC9A6基因突变小鼠小脑中可观察到浦肯野细胞的广泛进行性丢失及胶质化[14]。在MRI上,胶质增生表现为T2/液体衰减反转恢复序列上呈现的高信号,所以在Christianson综合征患者中,T2和液体衰减反转恢复序列高信号是第二常见的神经影像学表现。而在Angelman综合征中,MRI通常正常,或可能表现为轻度脑萎缩、髓鞘延迟、白质体积减少或局灶性白质信号改变,这是区别于Christianson综合征的影像学特征[13]。本文中先证者之兄的头颅MRI已经存在萎缩性改变,而先证者目前年龄为1岁11个月,尚需后期影像学检查随诊。
SLC9A6基因位于Xq26染色体上,编码一种Na+/H+交换蛋白(NHE6)。NHE6表达广泛,在可兴奋的组织,如大脑、心脏和骨骼肌中最为丰富,被认为参与了细胞内囊泡和突触囊泡的循环[5];在中枢神经系统中,NHE6在小脑、海马、皮质和浦肯野细胞层中表达得特别高,而在非神经细胞中,转运体的大部分优先累及在转铁蛋白受体富集的循环内小体中,它参与了囊泡pH值的调节和转运以及上皮细胞极性的维持[15]。所以Christianson综合征的变异性可能取决于NHE6突变的严重程度与其他pH调节溶质载体蛋白的功能重叠或遗传修饰效应,NHE6的功能缺失突变破坏了循环内小体的功能,最终导致神经元变性和细胞死亡。
Christianson综合征以X连锁方式遗传,对同胞的风险取决于母亲的遗传状况,与大多数X连锁疾病一样,杂合子雌性在每次妊娠中有50%的机会传播SLC9A6致病变异,所以对男性有明显的影响,而女性携带者则可能是无症状的,或者有轻微的表型。有研究结果表明女性携带者表型中并没有小头畸形[16],但可能存在学习困难、语言延迟、轻度至中度智力及行为问题,精神疾病的风险亦增高,包括抑郁症、焦虑症、多动症、强迫症等。本文中先证者的母亲智力正常,目前未有出现神经系统症状。但有文献报道随着年龄的增长,老年的女性携带者亦可出现神经退行性疾病,如帕金森病[17]。
本病尚无有效的治疗手段,主要采用支持和对症处理。本文2例患儿均有神经系统症状,给予抗癫痫药物控制发作及康复锻炼,但是最终的预后还需要长期随访。另外Christianson综合征在临床上与自闭症有重叠,相当一部分患有Christianson综合征的男孩曾接受过自闭症的诊断及治疗,本文中先证者之兄就曾接受自闭症的相关治疗,无明显效果。而分子遗传检测能够识别产生新的致病变异的家庭成员,避免早期过度医疗,也可以帮助确定遗传风险状况。
所有作者声明不存在利益冲突
None declared


























