
自身免疫性胶质纤维酸性蛋白星形胶质细胞病是一种近年来报道较罕见的免疫介导中枢神经系统炎性疾病,其特异性的生物学标志物为抗胶质纤维酸性蛋白抗体。文中就该疾病的病因发病机制、临床表现、辅助检查及治疗进行全面阐述,以提高广大临床医师,特别是神经专科医师对于该病的认识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2023 年从第1 期开始共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。本年度继教答题得学分活动于10月31日结束。
自身免疫性胶质纤维酸性蛋白星形胶质细胞病(autoimmune glial fibrillary acidic protein astrocytopathy,GFAP-A)是一种新型的中枢神经系统自身免疫炎性疾病,临床表现为脑膜、脑实质、脊髓、视神经、周围神经受累或上述各受累部位症状的组合。其中以急性或亚急性起病的脑膜脑炎最常见,并伴有前驱症状。胶质细胞酸性蛋白免疫球蛋白G(glial fibrillary acidic protein-IgG,GFAP-IgG)阳性是目前国际公认的GFAP-A诊断标准,且脑脊液阳性预测值高于其血清检测结果,推荐采用基于细胞学检测法(cell-based assay)或基于组织学检测法(tissue-based assay)。
2016年Lennon教授团队首次报道了16例GFAP-A患者的临床症状及影像学表现,包括炎性脑膜炎、脑炎和脊髓炎,并指出抗GFAP抗体是该病特异的生物学标志物[1]。先前有针对犬的研究已经报道了类似疾病,包括坏死性脑膜脑炎和肉芽肿性脑膜脑炎。2007年Shibuya等[2]证实患坏死性脑膜脑炎的犬脑脊液中存在抗GFAP抗体。GFAP是成熟星形胶质细胞的主要中间丝蛋白,具有参与维持细胞形态稳定、血-脑屏障形成、调节突触功能等多种生物学作用[3]。目前,国内外共报道GFAP-A患者500余例,报道较多的国家主要为美国、中国、意大利、日本、法国和英国[4, 5, 6, 7, 8, 9, 10]。
GFAP-A病因尚不明确,部分可能与感染、肿瘤相关。30%的患者存在前驱感染症状,常见流涕、咽痛、咳嗽等。还可发生在单纯疱疹病毒、水痘带状疱疹病毒染、人类免疫缺陷病毒、登革热和梅毒等感染后[4,8,11, 12],此外,GFAP-A与伴胼胝体压部可逆性病变的轻度脑炎相关的研究也支持其感染触发因素[13]。少数GFAP-A患者合并肿瘤,尤以卵巢畸胎瘤常见。此外,鼻咽癌、B细胞淋巴瘤、慢性淋巴细胞白血病及几乎所有器官的腺癌都有报道[4, 5, 6, 7, 8, 9, 10],推测肿瘤表达的GFAP可能触发副肿瘤性神经自身免疫[4]。GFAP-IgG为细胞内抗原抗体,自身并无致病潜力,淋巴细胞、小胶质细胞、巨噬细胞和浆细胞分泌的抗体相互作用所致的神经炎症是GFAP-A可能的发病机制[12]。
另外,约30%~40%的GFAP-A患者存在1种或多种神经元自身抗体,尤以抗N-甲基-D-天冬氨酸受体抗体IgG(NMDAR-IgG)最为常见,其次为抗水通道蛋白4抗体IgG(AQP4-IgG)、抗髓鞘少突胶质细胞糖蛋白抗体IgG(MOG-IgG),也有患者同时具有3种不同抗体(GFAP-IgG、AQP4-IgG、MOG-IgG)[4,14],少数可合并抗神经元核抗体-1、浦肯野细胞胞质抗体、富亮氨酸胶质瘤失活蛋白1抗体、接触蛋白相关蛋白2抗体和谷氨酸脱羧酶-65亚型抗体[4]。初始的免疫攻击致星形胶质细胞功能失调、释放趋化因子[肿瘤坏死因子、白细胞介素(IL)-1β、IL-6、IL-17],炎性细胞聚集可能在GFAP-A的发病机制中起重要作用[12]。
GFAP-A通常呈急性或亚急性起病,少数呈慢性起病,表现为进行性加重或复发缓解病程[15]。男女发病比例相当,好发于中年人,发病中位年龄为40~50岁,中位病程约12个月[4, 5, 6, 7, 8, 9, 10]。
临床初期表现为:头痛、颈强、流涕、咽痛、发热、乏力、咳嗽常见,多数患者最初被怀疑感染性脑膜脑炎[16]。
病程中常见的症候以脑症候为主[4, 5, 6, 7, 8, 9, 10],可表现神经精神症候。如意识障碍(5%~50%)、精神行为异常(9%~36%)、癫痫发作(11%~32%)、认知障碍(16%~30%)。认知障碍主要表现为记忆力下降、执行功能下降。此外,患者可以脑干脑炎为主要表现,如出现顽固性呃逆、眼球运动障碍、吞咽困难等;也可以脑膜炎为主要表现,这样的患者因为脑脊液常规白细胞计数增高、生化检查中葡萄糖及氯化物水平降低和蛋白增高,常常按结核性脑膜炎和(或)病毒性脑膜炎进行诊治,同时由于给予激素或静脉注射免疫球蛋白(IVIG)治疗使病情好转,而误以为诊断正确[17];还有被诊断为脊髓炎或播散性脑脊髓膜炎的情况发生。
GFAP-A患者也常有视神经症候(5%~63%)[4, 5, 6,10]。与AQP4-IgG阳性患者因视神经炎(optic neuritis)引起疼痛性视力丧失不同,大多数GFAP-A患者视力模糊与视盘水肿相关。多数患者无颅高压,视盘水肿可能是源于小静脉炎症导致的视神经乳头炎[4,18]。患者视觉诱发电位可见异常,眼科检查可发现双侧对称的视盘水肿,但严重视力损害罕见[18, 19],光学相干断层扫描显示视网膜神经纤维层增厚。少数患者视野检查可见轻度盲点扩大或弓形暗点。
运动障碍症候在GFAP-A患者中较常见,最近一项研究发现74/87(85%)的GFAP-A患者有运动障碍,包括共济失调(49%)、震颤(45%)、肌阵挛(37%)[20]。震颤表现为姿势性或动作性,多见于上肢。肌阵挛常发生于上肢和下肢。其他少见的运动障碍有:运动困难、眼阵挛、肌强直、肌纤维颤搐、舞蹈徐动症。也可表现为帕金森病。Tomczak等[21]报道1例66岁男性GFAP-A患者,3个月内出现进行性嗜睡、淡漠、焦虑、右臂震颤、尿潴留、进行性无力和跌倒,随后出现急性妄想性精神病,入院时体检显示有认知障碍、肌阵挛、强直、右手动作性震颤、运动迟缓和构音障碍,脑脊液蛋白及白细胞升高,MRI示大脑非特异性T2/FLAIR高信号和颈髓纵向广泛的横贯性脊髓炎。血清和脑脊液均证实为GFAP-A。无论罗匹尼罗还是卡比多巴/左旋多巴都不能改善该患者的帕金森病症状,而皮质类固醇治疗后其临床和影像学得到改善,但因激素依赖,患者接受了抗CD20抗体免疫治疗使得病情得到根本好转。
部分GFAP-A患者有周围神经系统受累(约24%),多表现为轴索大纤维多发性神经病、脑神经病变,其次为神经根神经病、感觉神经病[10]。Paul等[22]报道6例GFAP-IgG相关的炎性周围神经病患者,表型包括:不对称的近端无力(4/6),感觉性共济失调和长度依赖性感觉异常(1/6),神经性疼痛和长度依赖性无力(1/6);其中3例多发性神经根神经病患者具有脱髓鞘特征(1例传导速度减慢和潜伏期延长,另2例急性发作时F波延长或消失)。3例有轴索病变的患者远端肌肉存在纤颤电位。2例患者MRI显示周围神经强化。1例近端腓肠神经活组织检查证实了轴突变性和脱髓鞘以及神经外膜和内膜血管周围T细胞占优势的炎性聚集。亚急性、不对称、神经根或近端神经病变是典型的GFAP-A相关周围神经表现。
此外,GFAP-A患者还可有自主神经功能障碍(主要为排尿障碍)、眼震、感音神经性耳聋[23]、呼吸衰竭等表现。
并发症情况:患者住院期间常见的并发症是低钠血症,抗利尿激素异常分泌综合征可能是产生低钠的原因之一;其次是血栓栓塞[7]。20%以上的GFAP-A患者合并自身免疫病,包括1型糖尿病、自身免疫性甲状腺疾病、类风湿关节炎、溃疡性结肠炎、银屑病性关节炎[4,6]。
儿童GFAP-A的临床特点有以下几方面:Fang等[24]对35例儿童GFAP-A患者进行总结分析(包括男童23例,女童12例,男女比例约为2∶1),发病年龄(6.3±0.6)岁,最小为1岁2个月。临床表型与成人基本一致,表现为发热(62.9%)、头痛(42.9%)、抽搐(42.9%)、精神行为异常(51.4%)、意识障碍(54.3%)、视觉障碍(22.9%)、共济失调(11.4%)、瘫痪(40.0%)、自主神经功能障碍(25.7%)。1例患儿仅表现为周围性面瘫,1例表现为Bickerstaff脑干脑炎。11例(31.4%)患儿合并其他抗体(MOG-IgG、NMDAR-IgG、AQP4-IgG)阳性。儿童头颅MRI对比增强病灶主要位于脑膜和脑叶,室周线性增强比例儿童(8.5%)明显低于成人。尽管部分患儿有反复复发(17.1%)和遗留后遗症状(20%),大多数患儿预后良好。
总之,GFAP-A的临床表现可为脑膜、脑实质、脊髓、视神经甚或周围神经受累或上述各受累部位症状的组合,其中以脑膜脑炎最多见,临床上要注意鉴别。
90%以上的GFAP-A患者表现为炎性脑脊液,大于80%患者的脑脊液白细胞计数升高,表现为以淋巴细胞为主的白细胞升高(中位数为50~80个/μl),且淋巴细胞数增多可持续数月。脑脊液蛋白水平升高(>0.5 g/L),少数患者脑脊液葡萄糖含量降低,压力升高,约半数患者脑液寡克隆区带为阳性[4,16]。部分患者在发病后的第1个月内脑脊液腺苷脱氨酶(adenosine deaminase)水平短暂升高[7],注意要与结核性脑膜炎相鉴别。
1.头颅MRI:大多数GFAP-A患者头颅MRI检查异常,可累及大脑皮质、基底节区、脑室周围白质、下丘脑、脑干、小脑、脑膜以及颅骨等部位。多数情况下是侧脑室旁散在的细线样的在轴位上垂直于侧脑室的长T1、T2信号,FLAIR上是更明显的高信号,无明显占位效应,脑室周围病变恶化可进展为广泛性脑白质病变。Kimura等[7]报道最常见的异常信号位于基底节,其次是丘脑,认为双侧丘脑后部高信号是其特征性的表现。患者DWI通常是正常的。约半数患者头增强MRI异常,典型表现为从侧脑室向外延伸的血管周围线样放射状强化,常见淡淡的线状强化(图1A),容易被忽略,小脑也可有这种强化[1,4,16](图1B)。Mirian等[25]发现1例患者表现出从延髓和脑桥腹侧发出的线状放射样强化。其他还可有软脑膜强化(图1C)、点状强化、蛇形强化和室管膜强化[4]。
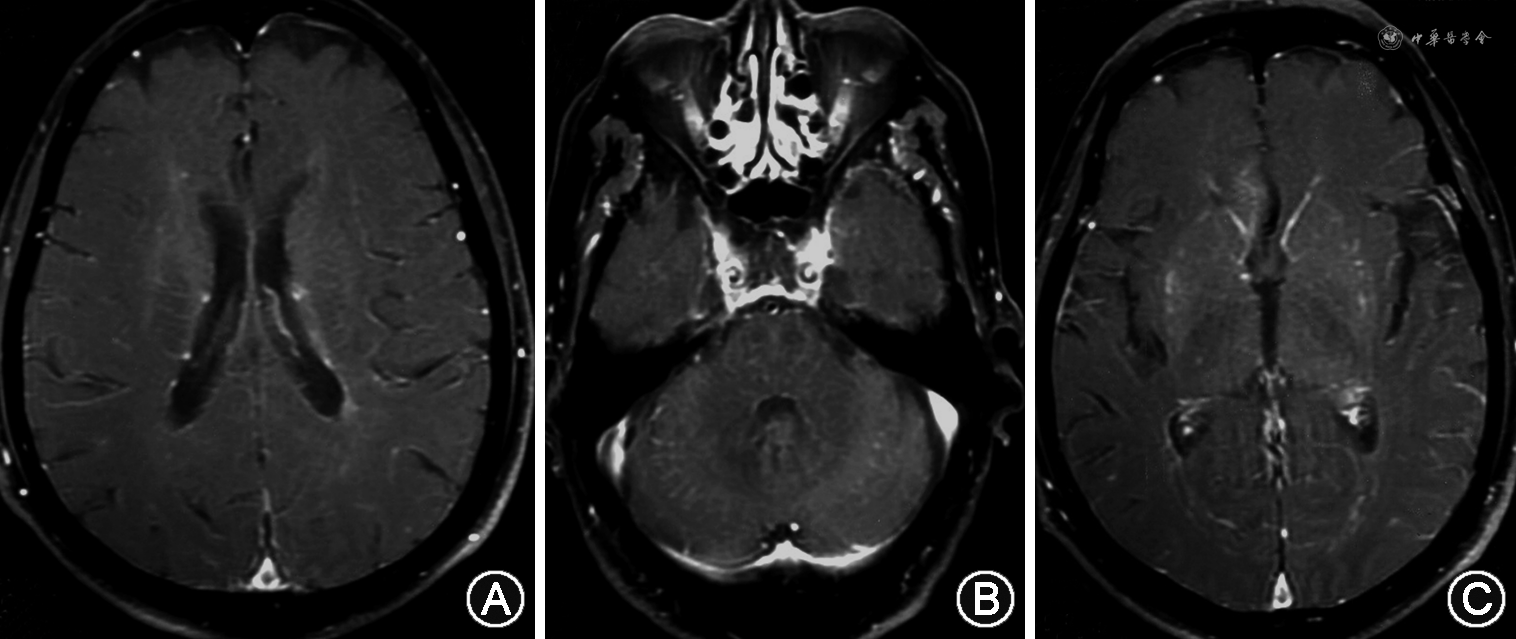
2.脊髓MRI:脊髓病变以颈胸髓为主,多数可累及3个及以上椎体节段,但与AQP4抗体阳性和MOG抗体阳性脊髓炎不同,其显影相对模糊,少有脊髓肿胀。增强扫描典型表现脊髓中央管、斑点样和脊膜强化[4],可与AQP4抗体阳性脊髓炎典型的斑片状、环形强化相鉴别。
3.氟代脱氧葡萄糖正电子发射计算机断层扫描:患者脑部病变部位可出现氟代脱氧葡萄糖摄取增加,支持弥漫性炎性反应[5,16]。
总之,GFAP-A的神经影像也是丰富多彩的,与脑膜脑炎,脑脊髓炎、视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorders,NMOSD)等有相似之处,但其强化表现有其特点,要注意仔细观察与甄别。
GFAP-A患者颅内病变累及脑膜和脑实质,脑膜可见CD8+T淋巴细胞、巨噬细胞和多核巨细胞为特征的炎性反应[6];颅内血管周围广泛的炎性细胞浸润,可见CD3+、CD4+、CD8+T淋巴细胞[4,27, 28, 29],也有报道可见CD20+B淋巴细胞[5];脑实质炎性浸润以CD8+T淋巴细胞占优势,小胶质细胞活化[29],部分存在AQP4和GFAP脱失,也可没有脱髓鞘和GFAP染色丢失的表现。
GFAP-A早期最容易被误诊为病毒性脑膜脑炎、结核性脑膜脑炎或细菌性脑膜炎等感染性疾病,往往采取抗病毒、抗痨或抗感染治疗。因其病因不明确,有的采取抗感染及免疫抑制等联合治疗。有的患者因为存在其他抗体,使得诊断更加模糊。为此,有必要重视疾病之间的鉴别诊断(表1)。

Differential diagnosis between autoimmune glial fibrillary acidic protein astrocytopathy(GFAP-A)and anti-myelin oligdendrocyte glycoprotein-IgG associated disorders(MOGAD)and neuromyelitis optica spectrum disorders(NMOSD)[4-10,30-34]
Differential diagnosis between autoimmune glial fibrillary acidic protein astrocytopathy(GFAP-A)and anti-myelin oligdendrocyte glycoprotein-IgG associated disorders(MOGAD)and neuromyelitis optica spectrum disorders(NMOSD)[4-10,30-34]
| 特征 | GFAP-A | MOGAD | NMOSD |
|---|---|---|---|
| 生物学标志物 | 脑脊液GFAP-IgG阳性 | 血清MOG-IgG阳性 | 血清AQP4-IgG阳性 |
| 发病年龄(中位数,岁) | 45 | 31(儿童常见) | 40 |
| 女∶男 | 1∶1 | 2∶1~1∶1 | 9∶1~8∶1 |
| 临床表现 | 脑膜脑炎、脑膜脑脊髓炎、癫痫、精神行为异常 | 复发性视神经炎,ADEM、脑炎或脑膜脑炎、视神经-脊髓炎 | 较严重视神经炎,LETM、极后区综合征,脑干综合征、急性间脑综合征、大脑综合征 |
| 脑炎比例 | 42.0% | 20.7% | 3.6% |
| 脊髓炎比例 | 10.0%~68.4% | 20%~30% | 90%~100% |
| 视神经病变 | 视乳头水肿多见 | 球后视神经炎多见、视乳头水肿 | 球后视神经炎 |
| 脑脊液细胞数增多 | 常见(>80%患者) | 常见(>70%患者) | 常见(>70%患者) |
| 脊髓MRI | 全脊髓均可累及,颈胸髓为主,病灶边缘不清晰,脊髓肿胀少,点状强化、中央管强化、软脊膜强化 | 长节段、不连续中央病灶,累及胸腰段及圆锥,脊髓强化、水肿少见 | 长节段,连续中央病灶,累及颈胸髓,脊髓多肿胀明显,云雾状、亮斑样、环形强化 |
| 脑MRI | 典型表现为皮质受累,从侧脑室向外延伸的血管周围线样放射状强化,脑膜强化 | 大片弥漫病灶,累及四脑室周围,小脑中脚,丘脑、脑桥病变更常见,16.3%皮质受累,6.1%脑膜病变 | 第三、四脑室,导水管周围T2高信号,极后区病变 |
| 视神经MRI | 多数未见异常 | 双侧受累,长病灶,视神经前段受累,视神经水肿明显,视神经鞘及周围脂肪组织强化具有特征性 | 双侧受累,长病灶,视神经后段或视交叉受累,中轴强化具有特征性 |
| 复发率 | 20%~50% | 40% | 90% |
| 预后 | 国外报道致残率低,国内报道多数遗留残疾 | 致残率低,发作后恢复较好 | 致残率高 |
注:GFAP-A:自身免疫性胶质纤维酸性蛋白星形胶质细胞病;MOGAD:抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G抗体相关疾病;NMOSD:视神经脊髓炎谱系疾病;GFAP:胶质纤维酸性蛋白;MOG:髓鞘少突胶质细胞糖蛋白;AQP4:水通道蛋白4;ADEM:急性播散性脑脊髓炎;LETM:纵向延伸的长节段横贯性脊髓炎;MRI:磁共振成像
1.NMOSD:NMOSD是一组自身免疫介导的以视神经和脊髓受累为主的中枢神经系统炎性脱髓鞘疾病。好发于女性患者,多以严重的视神经炎、长节段横贯性脊髓炎起病,部分以顽固性呃逆首发;头颅MRI典型的影像学表现为室管膜周围沿侧脑室、第三脑室、第四脑室室管膜,尤其靠近脑导水管出现T2WI高信号,也可出现广泛融合的白质病变,其强化以不均匀、边缘模糊的云雾状强化常见,该特点与GFAP-A不同。NMOSD急性期脊髓肿胀明显,可见不规则T1增强信号,而GFAP-A脊髓肿胀则少见[30]。NMOSD的视神经病变长于视神经的1/2,以视神经后段或视交叉受累多见,而GFAP-A仅表现为视盘水肿。AQP4-IgG是NMOSD高度特异性的诊断标志物,该疾病复发率高、致残率高,而GFAP-A相对复发少、致残率低。
2.抗髓鞘少突胶质细胞糖蛋白免疫球蛋白G抗体相关疾病(anti-myelin oligdendrocyte glycoprotein-IgG associated disorders,MOGAD):视神经炎是MOGAD最常见的首发临床表现,视盘水肿多见,眼眶MRI易累及视神经前段及视盘,50%的患者表现为视神经鞘和周围结构炎症。脊髓MRI影像学通常表现为长节段横贯性脊髓炎,强化和水肿发生率低,更易累及脊髓圆锥[31, 32, 33]。少见的MOG抗体相关性皮质脑炎,临床表现为癫痫发作(85%)、头痛(70%)、发热(65%)和皮质症状(55%)[34],GFAP-A与该疾病难以鉴别,需重视中枢神经系统自身免疫性疾病相关的抗体检测。
3.原发性中枢神经系统血管炎(primary angiitis of the central nervous system,PACNS):PACNS是一种主要累及脑、脊髓和软脊膜中小血管的中枢神经系统免疫性炎性疾病。一般缓慢起病,临床主要表现为头痛、认知功能下降、癫痫及局灶性神经功能缺损症状。病变多累及皮质、皮质下及深部白质,可呈带状、线状、多发团块状强化,因其血管易于破裂出血,SWI可呈低信号[35]。而GFAP-A颅内出血罕见。
4.神经元核内包涵体病(neuronal intraneuclear inclusion disease,NIID):GFAP-A需要与成人起病的NIID相鉴别。NIID患者首次就诊多在60岁左右,中枢神经系统症状包括发作性脑病、认知障碍、排尿障碍、肢体震颤、小脑共济失调、帕金森综合征、偏头痛发作等。其发作性脑病及双侧瞳孔缩小具有一定的临床诊断价值。典型头颅MRI表现为皮髓质交界区DWI高信号,部分患者可出现对称性脑白质病变、皮质肿胀和增强[36]。GFAP-A患者往往急性起病,其谵妄、意识模糊、认知障碍、震颤、癫痫等发作性脑病症状与NIID发作性脑病症状类似,故需要结合MRI影像表现对两者进行鉴别。
5.类固醇激素反应性慢性淋巴细胞性炎症伴脑桥血管周围强化症(chronicly mphocytic inflammation with pontine perivascular enhancement responsive to steroids,CLIPPERS):CLIPPERS是一种比较罕见的以淋巴细胞浸润为主、类固醇激素治疗有效的中枢神经系统慢性炎性疾病。MRI表现为以脑干和小脑为主的斑片状、放射状异常信号,增强后出现“胡椒粉样”弥漫斑点状、不规则线样强化。Yin等[37]报道1例患者反复发作性头痛7年,MRI示脑干、小脑、基底节多发曲线和点状强化,诊断为CLIPPERS,激素冲击治疗有效,但在减量过程中复发。最后1次发作时患者出现精神症状,查血清和脑脊液GFAP抗体阳性,最终诊断为GFAP-A。GFAP-A是否为CLIPPERS的临床结果之一仍有待探讨,因二者临床与影像学表现均有相似之处,长期随访及GFAP抗体检测有助于鉴别。
6.其他需要鉴别的疾病:自身免疫性脑炎、中枢神经系统淋巴瘤、胶质瘤、淋巴瘤样肉芽肿病等也需要与GFAP-A相鉴别。
关于GFAP-A的治疗目前尚无统一的指南或共识,急性期一线免疫治疗推荐大剂量糖皮质激素和(或)IVIG,重症患者需反复行血浆置换疗法[4, 5, 6, 7, 8, 9, 10]。20%~50%的患者有复发过程,需要长时间治疗[4, 5, 6, 7]。建议复发患者口服激素和免疫抑制剂,泼尼松60 mg/d或1 mg/·kg-1·d-1,总剂量不超过100 mg/d,连用3个月后再逐渐减量[12]。免疫抑制剂环磷酰胺、霉酚酸酯、利妥昔单抗似乎有更好的效果,而硫唑嘌呤在避免复发方面效果较差[8]。
总体来看,大多数GFAP-A患者预后良好,但国内外差别较大。梅奥诊所对38例患者进行中位时间20个月的随访,患者的改良Rankin量表(mRS)评分为1分[2],19例意大利患者经随访8.5个月,平均mRS评分为1.5分[6],法国对36例患者进行最短6个月的随访显示,患者的平均mRS评分为1分[10]。而国内学者报道多数GFAP-A患者遗留残疾且长期预后不佳[5,38]。
总之,GFAP-A在临床上可以说是一个超级模仿者。尤其在疾病早期表现为高热、头痛、认知障碍(谵妄、记忆力减退为主)、癫痫样发作,有脑膜刺激征,脑脊液表现与病毒、结核、细菌感染难以区分,容易被误诊为感染性脑膜脑炎。其次,可表现为视力异常、脑炎、脊髓炎、脑脊髓炎等相应症状,有时也伴有周围神经受累症状,需要与多种脱髓鞘病、自身免疫性脑炎等相鉴别。值得注意的是,GFAP-IgG阳性少数情况下可能是其他疾病的伴随情况,也有患者早期GFAP-IgG阴性,病情进展后复查出现阳性,要结合临床仔细甄别,必要时需多次复查GFAP抗体。对于存在多种抗体的情况时,一方面要从临床症候为主进行判断,另一方面要结合以抗体强阳性或滴度为主的一方进行综合判断。总之,对于GFAP-A患者尽早明确诊断,合理的免疫治疗及后续免疫修正治疗可减轻患者的病情进展,减少残疾并改善预后。
戚晓昆, 刁东卫. 自身免疫性胶质纤维酸性蛋白星形胶质细胞病[J]. 中华神经科杂志, 2023, 56(1): 82-87. DOI: 10.3760/cma.j.cn113694-20221002-00745.
所有作者声明无利益冲突
None declared
1.下面关于自身免疫性胶质纤维酸性蛋白星形胶质细胞病(GFAP-A)相关周围神经受累症状表述不正确的是?
A.亚急性起病
B.不对称的肢体无力
C.以神经根及近端神经病变为主
D.以远端神经病变为主
2.下列哪一项为GFAP-A自主神经功能障碍的主要表现?
A.体位性低血压
B.便秘
C.膀胱功能障碍
D.睡眠障碍
3.关于GFAP-A的典型头颅MRI表现,表述正确的是?
A.垂直于脑室的线样放射状强化
B.团块状强化
C.环形强化
D.斑片状强化
4.以下为GFAP-A常见病理表现的是?
A.血管周围广泛炎性细胞浸润
B.血管炎表现
C.白质脱髓鞘
D.血管增厚,较少炎性细胞浸润
5.GFAP-A最容易被误诊的疾病是?
A.视神经脊髓炎谱系疾病
B.感染性脑膜脑炎
C.抗髓鞘少突胶质细胞糖蛋白抗体相关疾病
D.神经元核内包涵体病

























