移植后淋巴组织增殖性疾病(PTLD)是与实体器官或造血干细胞移植相关的一组综合征,中枢神经系统PTLD(CNS-PTLD)非常罕见。文中报道1例肾移植术后24年的CNS-PTLD患者,病理学证实为弥漫大B细胞淋巴瘤,脑脊液病原学二代测序及病理学检查结果均支持Epstein-Barr病毒感染可能与之发生相关。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
移植后淋巴组织增殖性疾病(post-transplant lymphoproliferative disorder,PTLD)是接受实体器官移植或造血干细胞移植的患者在医源性免疫抑制的情况下发生的淋巴组织增殖的一系列综合征[1]。国外研究结果表明,PTLD的总发生率为0.47%,但受年龄与移植物影响较大,肾移植后PTLD的发生率为1%,小肠移植后的发生率高达20%,而在儿童患者中PTLD的发生率可能更高[2]。中枢神经系统PTLD(central nervous system-PTLD,CNS-PTLD)相对罕见,仅占15%[3]。我们报道1例CNS-PTLD患者并探讨该疾病的影像学与病理学特点。
临床资料 患者女性,59岁。主因“进行性右侧肢体力弱3个月,头晕、言语不利1个月”于2022年4月1日就诊于首都医科大学宣武医院。患者3个月前无明显诱因出现右下肢乏力,行走拖步,症状缓慢加重,逐渐出现右上肢力弱。1个月前出现持续性头昏沉感,同时言语不流利,忘记常用药物名称,睡眠增多,每日睡眠10余小时以上。无饮水呛咳,无构音不清,无复视或视力下降,无头痛,无恶心呕吐,无精神行为异常,无发热。患者自发病以来,饮食如常,无大小便障碍,无体重下降。患者有肾病病史30年,肾移植术后24年,目前服用他克莫司0.5 mg 2次/d,吗替麦考酚酯 0.25 g 2次/d,醋酸泼尼松片10 mg 1次/d及雷公藤多苷 10 mg 2次/d;患乙型病毒性肝炎10年,目前服用富马酸丙酚替诺福韦,病情控制稳定;患有2型糖尿病及2型糖尿病周围神经病。
专科体检:患者意识清醒,言语不流利,以运动性失语为主,记忆力、计算力轻度下降,理解力、定向力正常。脑神经检查无异常。右侧上下肢肌力Ⅳ级,左侧肢体肌力Ⅴ级。双上肢腱反射对称(++),双侧膝反射及跟腱反射消失,双侧病理征(+)。双下肢膝以下针刺觉减退。共济运动未见明显异常。行走右侧偏瘫步态。
生化检查:血常规:白细胞计数、血红蛋白含量及血小板计数均处于正常范围,中性粒细胞百分比76.3%(正常值50.0%~75.0%),淋巴细胞百分比15.6%(正常值20.0%~40.0%)。肌酐178 μmol/L(正常值78~108 μmol/L),尿素15.99 mmol/L(正常值1.70~8.30 mmol/L),乳酸脱氢酶254 IU/L(正常值109~245 IU/L),余肝功能及电解质均处于正常范围。感染八项:乙型肝炎表面抗原、乙型肝炎e抗原及乙型肝炎核心抗体均为阳性,乙型肝炎表面抗体及乙型肝炎e抗体为阴性,梅毒螺旋体特异性抗体、人获得性免疫缺陷病毒抗体及丙型肝炎病毒抗体均为阴性。肿瘤标志物:癌胚抗原5.31 ng/ml(正常值0~5.00 ng/ml),血清骨胶素CYFRA-21 4.65 ng/ml(正常值0.10~3.30 ng/ml),肿瘤相关抗原72-4 47.02 U/ml(正常值0~6.90 U/ml)。红细胞沉降率36 mm/L(正常值0~20 mm/L)。糖化血红蛋白6.8%(正常值4.0%~6.0%)。凝血功能+D-二聚体、尿常规、便常规、同型半胱氨酸、维生素B12及叶酸水平均处于正常范围。抗核抗体谱、抗磷脂抗体及抗中性粒细胞胞质抗体均为阴性。
腰椎穿刺脑脊液检验:脑脊液无色,清亮,压力110 mmH2O(1 mmH2O=0.009 8 kPa)。常规:白细胞计数16×106/L[正常值0~8×106/L],单核细胞百分比93.8%(正常值 60.0%~70.0%),多核细胞百分比6.2%(正常值30.0%~40.0%)。生化检查:蛋白定量0.96 g/L(正常值0.15~0.45 g/L),葡萄糖0.49 g/L(正常值0.45~0.80 g/L)、氯化物124 mmol/L(正常值118~128 mmol/L)。细胞学:镜下见多量淋巴细胞及少量单核细胞。脑脊液涂片革兰染色、墨汁染色及抗酸染色均阴性。脑脊液抗巨细胞病毒IgG抗体阳性,余TORCH抗体均为阴性。(血液+脑脊液)布鲁杆菌虎红试验未见异常。(血液+脑脊液)结核分枝杆菌游离核酸检测、莱姆病抗体、自身免疫性脑炎抗体、副肿瘤抗体、脱髓鞘疾病抗体及寡克隆区带均阴性。脑脊液流式细胞学:T细胞占脑脊液有核细胞41.3%,B细胞占脑脊液有核细胞0.2%,CD3-CD56+CD38+细胞占52.6%。脑脊液病原学二代测序Epstein-Barr病毒(Epstein-Barr virus,EBV)为阳性(序列数12)。
影像学检查:头颅CT(2022年4月1日)示左侧中脑、丘脑及基底节低密度影。头颅MRI(2022年4月2日)示左侧中脑、丘脑及基底节异常信号,累及对侧中脑及视束,强化后可见环形强化病灶(图1)。
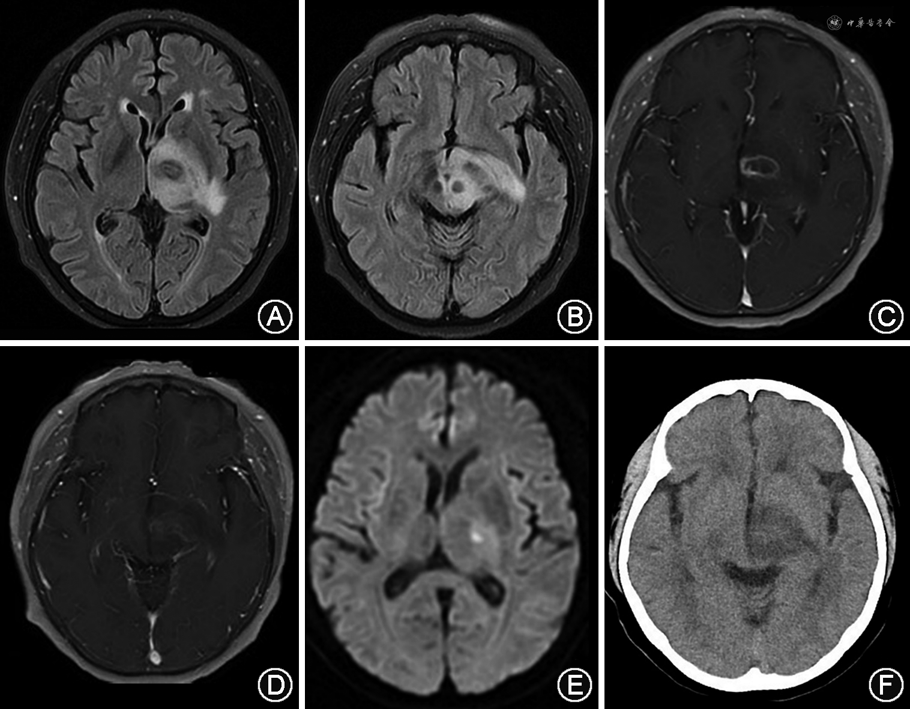
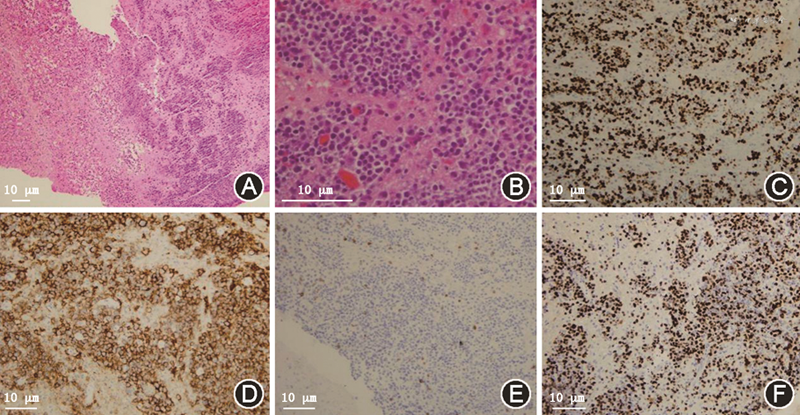
病理检查:患者为进一步明确诊断,2022年4月22日转入神经外科行“立体定向左侧基底节病变活组织检查术”。病理结果示:脑组织内异型细胞片状生长,部分围绕血管排列,核分裂象易见,伴有大片坏死。免疫组织化学:CD20(+),CD79α(+),CD3(-),CD5(-),CD30(+),B细胞淋巴瘤蛋白6(-),CD10(-),多发性骨髓瘤致癌蛋白-1(+),配对盒蛋白-5(+),Ki-67指数>70%,细胞周期蛋白D1(少量+),B细胞淋巴瘤蛋白2(+),P53(70%+),细胞性骨髓细胞瘤病毒癌蛋白(40%+),EBV编码RNA(原位杂交)(+)。临床病理诊断为:PTLD,EBV阳性弥漫大B细胞淋巴瘤,非生发中心型(图2)。
患者为中年女性,亚急性发病,病程为3个月。临床表现为进行性加重偏瘫,伴有头晕、言语不利及睡眠增多。既往有肾病、肾移植病史及乙型肝炎病史,长期应用免疫抑制剂。体检主要表现为运动性失语、高级皮质功能减退及右侧肢体偏瘫。影像学表现为左侧基底节、丘脑及双侧中脑占位性病变,脑脊液病原学二代测序发现EBV。综合进行性加重的临床表现、肾移植病史及颅内占位性病变情况,临床首先诊断考虑“PTLD”,鉴别诊断考虑机会性感染、胶质瘤及转移瘤。最终经病理诊断为PTLD。
目前已有大量研究支持EBV感染、免疫抑制状态以及遗传易感性在PTLD发病过程中起重要作用。EBV属于疱疹病毒家族,90%以上的成年人均处于EBV潜伏感染状态。EBV的主要宿主为B细胞,可同时感染部分T细胞、上皮细胞和肌细胞。在正常情况下,EBV感染的B细胞被宿主识别,通过CD8+细胞毒性T淋巴细胞对其进行杀伤和清除。同时,EBV能感染记忆性B细胞,产生潜伏膜蛋白从而上调抗凋亡基因的表达,且诱导B细胞进入潜伏感染状态,减少病毒抗原的表达,从而避免细胞毒性T淋巴细胞的杀伤。因此,EBV感染B细胞与宿主的免疫系统长期处于动态平衡的状态。在免疫抑制的情况下,T细胞功能被抑制,使得在人体内潜伏状态的EBV感染的B细胞逃脱免疫监视,出现B细胞异常增殖,这被认为是PTLD发病的根本原因[4]。本例患者脑脊液病原学二代测序发现EBV,病理学Epstein-Barr病毒编码RNA(EBER)原位杂交阳性,支持EBV与PTLD发生相关。但值得注意的是,既往已有EBV阴性PTLD的相关报道,发病时间通常在移植后数年,可能与长期免疫抑制剂应用有关,发病机制与EBV阳性PTLD可能有所差异[5, 6, 7]。
病理学诊断将PTLD分为早期病变、多形性PTLD、单形性PTLD及经典霍奇金淋巴瘤。早期病变是指浆细胞增生及传染性单核细胞增生症样病变,以大量免疫母细胞、浆母细胞及浆细胞弥漫增生而尚未完全破坏所累及组织及器官为特征。多形性PTLD是免疫母细胞、浆细胞和不同大小的淋巴细胞增生,浸润破坏淋巴结及结外器官,但尚不足以诊断淋巴瘤。而单形性PTLD则是指来源于单一克隆的细胞增殖,并且满足B细胞、T细胞或自然杀伤细胞淋巴瘤在一般人群中的诊断,常见弥漫大B细胞淋巴瘤与伯基特淋巴瘤。而经典霍奇金淋巴瘤则是PTLD的罕见类型。PTLD背景下的中枢神经系统淋巴瘤同样好发于幕上深层结构及脑室周围,较少发生在小脑和脑干[8],罕有脊髓和脊膜受累的报道。但是,PTLD、长期服用免疫抑制剂的其他疾病患者、获得性免疫缺陷综合征等与免疫功能正常的淋巴瘤在病理表现上亦有所区别。大多数免疫功能正常的患者肿瘤细胞罕见坏死及出血,且EBER原位杂交为阴性,而在免疫缺陷患者及部分免疫正常的老年患者中,坏死则是其常见的病理表现,出血的可能性亦高于大多数免疫功能正常患者,且通常有EBV感染的病理学依据[9, 10]。
在影像学方面,淋巴瘤肿块通常在CT上表现为等密度或高密度肿块,相应病变在MRI的T1与T2加权像上均为低信号或等信号,在DWI则为稍高信号,这是因为肿瘤细胞致密排列且核质比较高。免疫功能正常患者的淋巴瘤与PTLD的病理学差异,亦可在影像学有所体现。免疫功能正常患者强化MRI的特征性表现为均匀一致的实性强化,而免疫缺陷患者则因肿瘤的中心性坏死表现为环形强化,如合并出血,则T1加权像可能出现高信号,T2加权像相应为低信号[11, 12, 13]。
CNS-PTLD的临床表现与受累部位高度相关,可表现为头痛、恶心及呕吐等高颅压症状,亦可出现与病灶部位相关的神经功能缺损与认知障碍等。CNS-PTLD目前的主要治疗方案包括抗病毒、α干扰素、抗B细胞单抗、自体EBV特异性细胞毒性T细胞治疗、联合放射化学治疗及局部手术切除等,然而PTLD的疗效有限,大多预后不良。
综上所述,CNS-PTLD是罕见的移植后合并症,其诊断在临床上存在很大困难,应积极进行有创检查,提高诊断的准确率。该疾病目前没有标准的治疗方案,应根据患者移植后的病情、PTLD的部位及进展速度等综合选择。
崔博, 王丹丹, 朴月善, 等. 移植后中枢神经系统淋巴组织增殖性疾病1例[J]. 中华神经科杂志, 2023, 56(5): 549-552. DOI: 10.3760/cma.j.cn113694-20220928-00729.
所有作者声明无利益冲突
None declared


























