
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患儿男,7岁,也门籍,2015年7月为治疗双侧眼球突出、改善闭眼功能入院。患儿出生后即被发现身体明显异常,头部和四肢呈明显畸形外观,随年龄增长,患儿身体及智力发育明显滞后。在1~2岁之间,曾先后3次就诊埃及某公立医院,全麻下行唇裂修复术、腭裂修复术、额骨开窗术(具体不详),术后恢复良好,但遗留口唇运动障碍、头颅继发畸形等。患儿系第4胎,足月顺产,父母既往体健,近亲结婚(堂兄妹),4名子女,3女1男,其中第2胎女孩也表现为相似的畸形外观。父母否认遗传性疾病史及妊娠期药物接触史。2009年经埃及艾因·夏姆斯大学遗传医学中心外周血染色体检查,诊断为Roberts综合征。检查:患儿呈明显的异常发育外观,身高62 cm,体重7 kg,可站立,行走吃力,步态蹒跚,目光略显呆滞,言语不清,查体欠合作。头部外形异常,毛发稀疏,头颅前后径大于左右径,颅顶正中冠状位可见明显的骨性挛缩带,额部中线区域皮肤软组织规律性搏动,触之柔软。双侧眼球明显突出,眼睑闭合不全,由于患儿头颅横径过小,常规眼球凸度计无法测量。面中部平坦,无明确鼻小柱及鼻翼结构。上唇突出,皮肤可见手术遗留切口瘢痕,其舌侧与上牙槽骨紧密粘连,无法运动,口唇闭合不能,口内牙列排列异常。双上肢"海豹肢"畸形,双手均为4指,伴有并指畸形。双下肢短小,均为屈曲状态,膝关节无运动功能。会阴及外生殖器未见明显异常。超声心动图检查,提示先天性心脏病(动脉导管未闭)。颅脑CT检查,提示冠状缝分离,额骨缺损,多处见金属伪影。此外,CT及MRI均提示颅脑为宫内感染后遗表现。四肢X线检查,提示双上肢肱骨及尺、桡骨缺如,3根掌骨远端与指骨相连;双下肢无胫、腓骨结构,长骨于膝关节处成角融合(图1)。
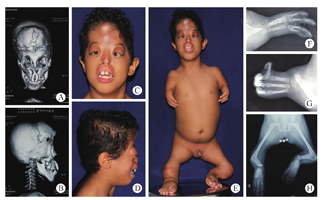
患儿入院后,经院际间多学科(整形外科、神经外科、麻醉科、儿科、眼科、口腔科、检验科等)联合会诊后,拟于全麻下行"颅腔开大、颅骨重排、额眶带迁移手术"。但因患儿体质虚弱,入院不久便出现严重的上呼吸道感染症状及体征,遂出院回国休养,后失联。
Roberts综合征是一种罕见的常染色体隐性遗传疾病,其特征性临床表现包括出生前后的发育迟滞、颅颌面畸形、短四肢畸形、智力障碍等[1]。1919年由Roberts[2]首次报道,50年后Herrmann等[3]又报道了非常相似的病例,并将其命名为SC短肢畸形综合征。目前,已明确这2种综合征系同一种疾病不同程度的表现形式,前者的畸形情况更为严重[4]。除了特征性的临床表现外,标准的细胞遗传学检查对于诊断该类疾病非常重要。2005年,Vega等[5]证实Roberts综合征是由于位于8p21.1染色体上的ESCO2基因突变所致。根据临床表现及相关的实验室检查本例患儿可明确诊断为Roberts综合征。
通常,伴有严重畸形的个体往往会发生流产或产后早期死亡,而畸形较轻的则可存活至成年阶段[6],成年后因心肺功能衰竭、肿瘤等原因,患者的死亡率也会增加[3,7]。到目前为止,国际上仍缺乏关于这类疾病成年患者的远期观察随访[4]。
有学者认为,应该积极地给予患儿必要的医疗救治,这不仅能够改善畸形外观,重建器官功能,也能更好地为临床提供相关的愈后信息[8],我们也十分赞同这个观点。但是,本例虽然进行了多次的颅面及唇腭裂修复手术,仍存在严重问题:①颅腔仍严重狭窄,限制大脑继续发育;②额眶带前移效果微弱,致使眼球仍突出明显;③唇裂纠正不足,上唇与上颌骨贴合,严重限制口唇运动。由于该类患儿发育极差,常合并严重心、肾异常,因此,每一次麻醉及手术创伤均可能危及生命。作为外科医生,我们有必要更好地认知此综合征,并在合理的评估下联合相关科室给予相应的手术治疗。

























