
肯尼迪病(Kennedy disease,KD)又称脊髓延髓肌萎缩症(SBMA),是一种罕见的X染色体连锁隐性遗传的下运动神经元变性疾病,发病机制为雄激素受体(AR)基因1号外显子编码多聚谷氨酰胺的CAG异常扩增[1]。重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经肌肉接头传递障碍的自身免疫性疾病,乙酰胆碱受体(AChR)抗体(AChR-Ab)为最常见的致病性抗体,可干扰AChR聚集、影响AChR功能及神经肌肉接头信号的传递[2]。KD与MG都属于神经肌肉罕见病,均可出现四肢无力和延髓麻痹等症状。笔者现报道河北以岭医院重症肌无力科收治的1例KD合并AChR-Ab阳性型MG的老年男性患者,总结其治疗经验,以期为临床作者提供参考。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肯尼迪病(Kennedy disease,KD)又称脊髓延髓肌萎缩症(SBMA),是一种罕见的X染色体连锁隐性遗传的下运动神经元变性疾病,发病机制为雄激素受体(AR)基因1号外显子编码多聚谷氨酰胺的CAG异常扩增[1]。重症肌无力(myasthenia gravis,MG)是由自身抗体介导的获得性神经肌肉接头传递障碍的自身免疫性疾病,乙酰胆碱受体(AChR)抗体(AChR-Ab)为最常见的致病性抗体,可干扰AChR聚集、影响AChR功能及神经肌肉接头信号的传递[2]。KD与MG都属于神经肌肉罕见病,均可出现四肢无力和延髓麻痹等症状。笔者现报道河北以岭医院重症肌无力科收治的1例KD合并AChR-Ab阳性型MG的老年男性患者,总结其治疗经验,以期为临床作者提供参考。
患者 男性,68岁,因间断四肢无力、构音不清、咀嚼无力5年,再发加重2个月,于2021年6月入河北以岭医院重症肌无力科。患者于2016年3月无明显诱因出现双眼睑下垂,症状呈波动性,晨轻暮重,劳累后加重,休息后减轻;8月患者劳累后出现双眼睑下垂加重,同时伴有咀嚼无力、饮水呛咳、构音不清、气短、四肢无力等症状,就诊于当地市医院,行新斯的明试验阳性,诊断"MG";2020年12月曾入我院,给予胆碱酯酶抑制剂、激素治疗10 d,症状好转出院。出院3个月后停服激素,以每日60 mg溴吡斯的明维持,5年来饮食吞咽正常,言语清晰,四肢有力,无胸闷气短等症状。患者2021年4月劳累后出现鼓腮无力,说话不清,咀嚼、吞咽困难,四肢无力,于6月再次就诊于我院。患者已婚,50岁左右性功能丧失,育1子1女,均体健,否认家族成员中有类似症状。既往乳腺增生10余年。体格检查:意识清楚,言语不清,定向力、记忆力、计算力正常;闭目不紧,埋睫征不全,眼裂等大,上眼睑未见下垂,双侧瞳孔正大等圆,对光反射灵敏,眼球活动灵活,无复视;鼓腮漏气,双侧咬肌无力,咽反射消失,伸舌居中,舌肌萎缩,中央有纵行沟槽(图1A),口周肌肉萎缩(图1B),并可见细小肌束震颤。双侧乳房发育(图1C)。双手震颤,四肢近端肌肉轻度萎缩并可见粗大震颤,前臂肌群及胸肌可见细小束颤。颈部肌力4级,双上肢肩肘腕指肌力为4-4-4-4级,髋膝踝趾肌力为3-4-4-4级,四肢肌张力降低。肱二、三头肌腱反射减弱,跟、膝腱反射消失,霍氏征(-),巴氏征(-),腹壁反射消失,深浅感觉未见异常,肌疲劳试验阳性。MG定量评分(QMGS)为15分,MG绝对评分(ARS-MG)为28分,MG日常生活评分(MG-ADL)为12分,美国重症肌无力基金会(MGFA)分型标准为ⅣB型。
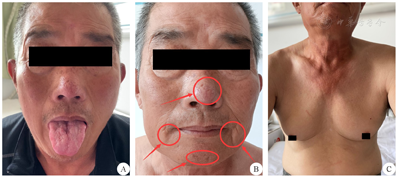
A:舌肌萎缩;B:口周肌肉萎缩;C:乳房发育
辅助检查:AR基因1号外显子CAG重复数为43(图2),AChR-Ab>20 nmol/L(↑),肌酸激酶371 U/L(↑),低密度脂蛋白胆固醇3.17 mmol/L(↑),总胆固醇5.71 mmol/L(↑),同型半胱氨酸25.08 μmol/L(↑),α-羟丁酸脱氢酶220 U/L(↑),γ-谷氨酰转肽酶10.7 U/L(↓),游离甲状腺素23.4 pmol/L(↑),促黄体素10.53 mIU/mL(↑)。血常规、尿常规、粪常规、睾酮、雌二醇、促卵泡素、孕酮、催乳素、肿瘤全项等未见异常。新斯的明试验阳性。肌电图:重复神经电刺激(RNS)低频波幅衰减明显,高频无递增(左副神经低频衰减33%,右副神经低频衰减38%,左面神经低频衰减26%,右面神经低频衰减16%);轻收缩时运动单位电位时限增宽,波幅增高,重收缩时运动单位电位募集减少,波形呈单纯相;感觉神经传导速度(SCV)减慢,波幅减低(右正中神经45.2 m/s、左尺神经24.1 m/s、右腓肠神经26.3 m/s;正中神经左侧4.5 μV、右侧1.4 μV,尺神经左侧5.3 μV、右侧1.4 μV,腓肠神经左侧1.9 μv、右侧1.5 μV,腓总神经左侧1.7 μV、右侧1.1 μV);运动神经传导速度(MCV)、F波及H反射潜伏期未见异常。胸部CT:右肺上叶尖后段实性小结节;左肺上叶小肺泡融合;支气管炎,左肺下叶背段轻度炎症;考虑胸腺增生。头颅MR:左额部伪影;双侧丘脑基底节多发腔隙性脑梗死,较陈旧;双侧侧脑室周围及双侧额顶叶多发小缺血灶;老年性脑改变。AR基因1号外显子CAG重复数大于38即可诊断KD;AChR-Ab阳性(大于0.45 nmol/L)且伴有典型特征即可诊断MG,本例患者符合KD与MG诊断标准。本例患者拒绝再行检查,遗憾没有预后对比。

KD发病率约为1/40 000,发病年龄为30~60岁[3,4]。正常人AR基因1号外显子的CAG重复数为10~36,而KD患者为38~62,目前基因检测CAG数>38即可确诊[3,5]。KD的临床表现以下运动神经元损害为主,其特征为缓慢进展的延髓、面部及四肢肌肉无力和萎缩[4]。最常见的症状为肢体无力,通常从近端开始,可伴有腱反射减弱或消失[6];其次为延髓功能障碍,可出现构音障碍、咀嚼吞咽困难和舌肌萎缩等;80%的患者会出现震颤,且多为姿势性手部震颤,同时伴有肌束颤动,尤其以口周和舌头最为明显[7,8];部分患者有乳房发育、睾丸萎缩、生育能力降低等,国内报道男性乳房发育发生率为34.8%,而AR基因异常的女性携带者一般无症状[7,9];患者死亡和住院的主要原因是吸入性肺炎[8]。大多数KD患者血清肌酸激酶水平升高,部分患者甘油三酯、低密度脂蛋白、丙氨酸转氨酶、天冬氨酸转氨酶升高[9,10]。肌电图以慢性神经源性损害为主,多伴感觉和运动神经传导异常,感觉神经动作电位波幅降低是KD特有表现[11]。有研究表明CAG重复数大于47时运动神经动作电位波幅下降明显,小于47时感觉神经动作电位波幅下降显著[12]。本例患者四肢、延髓肌无力症状明显,并伴有乳房发育、双手震颤与肌束震颤;肌酸激酶、低密度脂蛋白、总胆固醇升高;神经电生理检查示神经源性损害,SCV减慢,波幅减低;AR基因1号外显子CAG重复数为43,因此KD诊断成立。
我国MG发病率约为0.68/10万,各年龄段均可发病[13];具有典型临床特征,且满足药理学检查、血清AChR-Ab等检测、电生理学特征中的任意一项阳性即可诊断[2]。MG的主要临床表现为骨骼肌波动性无力和易疲劳,症状呈晨轻暮重,活动后加重、休息后可减轻。最常见的首发症状为上眼睑下垂和(或)复视,发病早期可单独出现眼外肌、咽喉肌或肢体肌肉无力;面肌受累可致眼睑闭合无力、鼓腮漏气等;延髓肌受累可致咀嚼吞咽困难、构音障碍、声音嘶哑及饮水呛咳;肢体无力以近端为著;约80%的患者伴有胸腺异常;主要死亡原因包括呼吸衰竭、肺部感染。AChR型MG少见肌肉萎缩,舌肌萎缩在肌肉特异性受体酪氨酸激酶(MuSK)型中较多见。药理学检查新斯的明试验多呈阳性;血清抗体检测包括AChR-Ab、MuSK抗体(MuSK-Ab)、低密度脂蛋白受体相关蛋白4抗体(LRP4-Ab)等,其中AChR-Ab阳性可达85%~90%;低频(2~3 Hz)RNS波幅衰减在10%以上[2]。本例患者早期出现眼睑下垂,继而出现四肢、延髓肌无力,症状呈波动性,晨轻暮重;新斯的明试验阳性;AChR-Ab检测阳性;低频RNS波幅衰减明显,最高衰减达38%;胸部CT提示胸腺增生,因此MG诊断成立。
KD为基因异常的下运动神经元病,MG为神经肌肉接头障碍的免疫性疾病,二者临床症状具有相似性,但在发病机制、诊断依据方面有明显差异。本例患者检查结果明确,同时患有两种疾病实属罕见。详细追问病史,患者约10年前出现双侧上臂肌束震颤,同期出现乳腺增生,这可能是KD的早期症状,医生与患者均未重视,也未进一步做肌电图与基因检查,5年前陆续出现眼睑下垂、延髓麻痹、四肢无力等症状。首次就诊时诊断为MG,但依据临床症状出现的顺序,KD应发病在先,MG发病在后,MG可能继发于KD,笔者推测两者有着共同的发病机制:(1)神经肌肉接头异常:既往研究表明运动神经元和肌肉中的雄激素及AR可改变神经肌肉接头突触前和突触后的特化形态,在调节神经肌肉接头功能中发挥作用,所以推测AR基因在KD中的异常可能导致神经肌肉接头功能障碍。(2)免疫系统异常:有研究表明雄激素可调节AChR的数量,雄激素的紊乱与MG的发病也有一定关系[14],推测异常的AR基因可能是通过改变AChR的数量从而导致异常的免疫应答,生成致病性AChR-Ab进而导致了MG。(3)基因异常直接导致:基因异常与免疫性疾病息息相关,不排除AR基因异常也可能是导致MG的直接原因,而两者之间是否有直接联系还有待进一步证实。当然,两种疾病集于一身也有可能是一种偶然现象。
KD迄今为止没有标准的治疗方案,多数临床试验研究是以抑制雄激素水平为目标,如应用亮丙瑞林、度他雄胺、克伦特罗等,但临床疗效不佳[15]。MG的治疗现以胆碱酯酶抑制剂、糖皮质激素、免疫抑制剂、静脉注射免疫球蛋白、血浆置换及胸腺切除等为主[2]。本例患者首次诊断为MG,给予溴吡斯的明、甲泼尼龙治疗后长达5年无自觉症状,5年后病情复发严重,给予静注人免疫丙种球蛋白、溴吡斯的明及甲泼尼龙治疗,1个月后构音障碍与肌无力症状明显改善,肌束颤动频率减少,但咀嚼功能恢复不明显,舌肌萎缩、手部震颤、乳房发育等症状无明显变化,评估QMGS为7分,ARS-MG为12分,MG-ADL为5分;4个月后随访,患者状态良好,双上肢肩肘腕指肌力为4-5-5-5级,髋膝踝趾肌力为4-5-5-5级,肌力基本正常,肌束颤动与手部震颤较前减轻,构音较前清晰但略显鼻音,咀嚼吞咽明显好转,但舌肌萎缩、乳房发育症状无变化,评估QMGS为4分,ARS-MG为6分,MG-ADL为2分,后期疗效需进一步随访观察。国外有报道3例KD伴有肌无力样症状,1例KD合并眼肌型MG,此4例患者低频RNS波幅有不同程度衰减,但均无血清免疫学抗体证据支持,治疗上均使用了溴吡斯的明,其中2例联合了糖皮质激素,症状均得到改善[16,17];国内有报道1例KD患者在使用糖皮质激素后肌无力症状可间断性减轻[18]。本例患者两次应用MG诊治方案均有明显疗效,这可能受益于积极的免疫干预治疗,而应用于MG的药物是否对KD有效还有待进一步证实。
所有作者均声明不存在利益冲突

























