
对利用生物信息学方法筛选的肾上腺皮质癌(ACC)肿瘤微环境(TME)相关基因构建的预后模型进行验证,为ACC的诊疗提供临床指导和相关生物标志物。
从癌症基因组图谱(TCGA)数据库中收集79例ACC患者的转录组数据和临床病理数据。采用ESTIMATE算法计算免疫评分、基质评分(二者即反映TME)和ESTIMATE评分,应用VennDiagram包对免疫评分及基质评分高、低评分组(以中位值进行分组)间差异表达基因进行选择,采用基因本体(GO)数据库和京都基因与基因组百科全书(KEGG)数据库对选择的基因进行功能富集分析,探索基因潜在功能与通路。采用单因素Cox回归、lasso回归和多因素Cox回归分析筛选与ACC TME相关的基因,并建立ACC患者预后的风险评分(RS)模型,用受试者工作特征(ROC)曲线评价构建的模型预测ACC患者预后的价值。以基因表达综合(GEO)数据库的数据集GSE33371和GSE19750作为外部验证集,对建立的预后模型进行验证。从TCGA数据库中提取79例ACC患者资料,将临床病理因素与所构建预后模型的RS纳入Cox回归分析,获得ACC患者预后影响因素。
根据免疫评分和基质评分,用VennDiagram包筛选得到1 205个二者交集的差异表达基因,其中上调833个,下调372个。对差异表达基因进行各回归分析筛选后,最终构建成功包含9个TME相关基因(GREB1、POU4F1、HIC1、HOXC9、CACNB2、RAB27B、ZIC2、C3、CYP2D6)的ACC预后模型,即RS=GREB1×0.223 6+POU4F1×0.671 7+HIC1×0.167 5+HOXC9×0.211 3+CACNB2×0.156 0+RAB27B×0.956 5+ZIC2×0.582 7+C3×(-0.003 1)+CYP2D6×0.819 3。该模型在TCGA数据库中预测79例ACC患者1、3、5年总生存的ROC曲线下面积(AUC)分别为0.876、0.919、0.917,在GEO验证集中预测45例ACC患者1、3、5年总生存的AUC分别为0.689、0.704、0.708,表明模型对ACC患者生存具有较高的预测准确性。对TCGA数据库中79例ACC患者资料进行Cox回归分析显示,TME相关基因预后模型RS是ACC患者预后的独立影响因素(HR=1.011,95% CI 1.005~1.016,P<0.01)。
构建的ACC TME相关基因预后模型可用于预测ACC患者的预后。模型中包含的9个基因有可能作为研究ACC发病机制和免疫治疗的新靶点,值得进一步研究。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肾上腺皮质癌(ACC)是一种较为罕见的恶性肿瘤,发病率为(1~2)/100万[1]。由于该疾病早期发病特征不明显,但恶性程度高、进展快,许多患者就诊时就已出现肿瘤浸润周围组织或远处转移。目前手术仍是局限性ACC的一线治疗方法,晚期患者可采用多学科综合疗法进行治疗,以达到改善预后的目的,但治疗效果均不理想[2]。因此,探究ACC发病的分子基础、研究ACC的基因特征,有助于研发更特异和有效的治疗方法[3]。肿瘤微环境(TME)不仅包括肿瘤细胞本身,还包括周围的成纤维细胞、免疫细胞、炎性细胞、胶质细胞等,以及浸润在邻近区域的细胞间质、脂肪细胞、微血管和生物分子[4]。目前,大量研究关注TME与生存结局或复发之间的关系。探索ACC与TME的相互作用有助于揭示促进肿瘤进展的机制,并可能为ACC治疗提供潜在的新靶点。近年来,随着基因组学的发展以及基因组学和生物信息学分析方法的广泛应用,研究人员能够从基因水平探究多种癌症的基因表达和表观遗传学改变。本研究采用ESTIMATE算法分析ACC的TME,计算免疫评分、基质评分、ESTIMATE评分,筛选与ACC预后相关的TME基因,建立预后模型,并与ACC患者的其他临床病理特征比较,验证模型的预后预测应用价值。
ACC患者转录组数据来自癌症基因组图谱(TCGA)数据库(https://portal.gdc.cancer.gov/),数据下载时间为2020年10月15日。检索条件:(1)知识库;(2)病例选择:原发位点为肾上腺,程序为TCGA,项目为TCGA-ACC,其余选项默认系统设置;(3)文件选择:数据分类为转录组分析,数据类型为基因表达量化,工作流类型为HTSeq-FPKM,其余选项默认系统设置。下载79例ACC患者的转录组数据。
ACC患者临床数据来自TCGA数据库,数据下载时间为2020年10月15日。检索条件:(1)知识库;(2)病例选择:同转录组数据"病例选择"条件;(3)文件选择:数据分类为临床,数据格式为bcr xml,其余选项默认系统设置。从下载得到的数据中提取具有转录组信息患者的临床数据,得到79例ACC患者的临床数据。提取的临床数据包括总生存时间、年龄、性别、临床分期、肿瘤大小、淋巴结转移、远处转移。总生存时间定义为从患者诊断至因任何原因死亡或数据下载的时间。
采用R语言软件ESTIMATE包计算得到79个样本的免疫评分、基质评分(二者即反映TME)和ESTIMATE评分[5]。分别以免疫评分、基质评分、ESTIMATE评分中位值为阈值,将患者分为高评分(≥中位值)组和低评分(<中位值)组。从基因表达综合(GEO)数据库下载GSE33371和GSE19750芯片数据集,采用R语言软件sva包对芯片数据进行批次校正,将该数据作为外部数据集,用于验证所构建的预后模型的预测效果。
用R语言软件Limma包对免疫评分或基质评分高、低评分组的转录组数据进行统计分析,采用wilcox.test检验筛选两评分高、低评分组间差异表达基因(DEG)[6];筛选标准:错误发现率(FDR)<0.05,|log[差异倍数(FC)]| ≥1。应用VennDiagram包对免疫评分和基质评分中的DEG取交集,作为候选基因。
应用R语言软件org.Hs.eg.db包、clusterProfiler包、enrichplot包和ggplot2包对候选基因进行基因本体(GO)数据库分析和京都基因与基因组百科全书(KEGG)数据库分析。P<0.05为差异有统计学意义。
提取GEO数据库中经免疫评分和基质评分筛选的DEG,应用R语言软件survival包进行单因素Cox回归分析,筛选标准为P<0.05。应用survival包对候选基因进行生存曲线绘制,筛选P<0.05的基因。应用survival包和glmnet包对候选基因进行lasso回归分析,对获选基因进行双向逐步多因素Cox回归分析,构建预后模型:风险评分(RS)=[∑(βi)]×[expi](i为预后基因个数)。应用survival包绘制高、低RS组Kaplan-Meier总生存曲线,绘制受试者工作特征(ROC)曲线,内部验证该模型预测预后的准确性。
统计分析和图形绘制采用R语言软件(4.0.2版)。正态分布计量资料以均数±标准差( ±s)表示,非正态分布计量资料以中位数(范围)表示;多组间非正态分布计量资料比较采用Kruskal-Wallis方差检验。以RS的中位值为阈值,将患者分为高评分(≥中位值)组和低评分(<中位值)组,用Kaplan-Meier法绘制免疫评分、基质评分、ESTIMATE评分、RS高评分组、RS低评分组总生存曲线,并行log-rank检验。应用R语言软件的survival包对从TCGA数据库中提取的ACC患者RS和临床病理因素进行单因素和多因素Cox回归分析,筛选影响ACC患者预后的独立危险因素。以P<0.05为差异具有统计学意义。
±s)表示,非正态分布计量资料以中位数(范围)表示;多组间非正态分布计量资料比较采用Kruskal-Wallis方差检验。以RS的中位值为阈值,将患者分为高评分(≥中位值)组和低评分(<中位值)组,用Kaplan-Meier法绘制免疫评分、基质评分、ESTIMATE评分、RS高评分组、RS低评分组总生存曲线,并行log-rank检验。应用R语言软件的survival包对从TCGA数据库中提取的ACC患者RS和临床病理因素进行单因素和多因素Cox回归分析,筛选影响ACC患者预后的独立危险因素。以P<0.05为差异具有统计学意义。
根据纳入的TCGA数据库中79例ACC患者的基因表达谱和临床数据计算,免疫评分、基质评分、ESTIMATE评分中位值分别为-190.51分(-1 179.24~1 980.65分)、-842.68分(-1 642.41~1 495.87分)、-942.77分(-2 766.65~3 441.57分),依据各评分中位值分组,各评分的高评分组均为39例,低评分组均为40例。各评分分组的Kaplan-Meier生存曲线显示,高、低免疫评分组ACC患者总生存差异有统计学意义(P<0.05)(图1A)。高、低基质评分和ESTIMATE评分组间总生存差异均无统计学意义(均P>0.05)(图1B、图1C)。临床分期Ⅰ~Ⅲ期患者免疫评分、基质评分和ESTIMATE评分均逐渐降低,Ⅰ~Ⅳ期患者间免疫评分和ESTIMATE评分差异均有统计学意义(均P<0.05),但基质评分差异无统计学意义(P>0.05)(图2)。
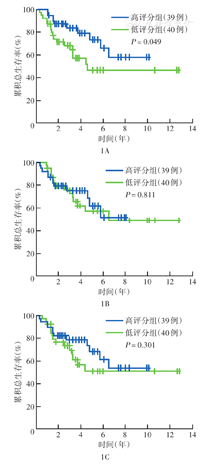
注:患者为从癌症基因组图谱数据库中纳入的肾上腺皮质癌患者;依据ESTIMATE包计算的各评分中位值分为高评分(≥中位值)组和低评分(<中位值)组;免疫评分、基质评分、ESTIMATE评分的中位值分别为-190.51分、-842.68分、-942.77分
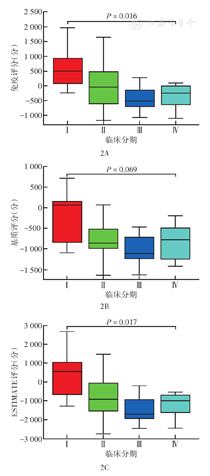
注:患者为从癌症基因组图谱数据库中纳入的肾上腺皮质癌患者;各评分依据ESTIMATE包计算获得;临床分期Ⅰ~Ⅳ期患者分别有9、38、16、16例
根据免疫评分,筛选出2 193个DEG,其中上调1 099个,下调1 094个。根据基质评分,筛选出1 559个DEG,其中上调1 021个,下调538个。对上述基因取交集得到1 205个免疫评分和基质评分交集基因,其中上调833个,下调372个。
对交集基因进行GO数据库功能富集分析结果显示,生物学过程中DEG主要参与免疫应答、淋巴细胞介导和白细胞调控等;细胞成分中DEG主要定位在免疫球蛋白、主要组织相容性复合体(MHC)Ⅱ类蛋白质复合物等;分子功能中DEG主要涉及抗原结合、免疫受体活动、免疫球蛋白绑定等。对交集基因进行KEGG数据库功能富集分析结果显示,主要的信号通路包含受体相互作用、趋化因子信号、病毒蛋白与细胞因子和细胞因子受体的相互作用、吞噬体、B细胞受体信号通路等。
免疫评分和基质评分中的1 205个DEG中,有684个存在于GEO数据集中。采用survival包,对684个候选基因进行单因素Cox回归分析,筛选出与预后相关的121个差异基因。通过绘制差异基因患者的Kaplan-Meier生存曲线,筛选出生存曲线中P<0.05的49个基因。对49个差异基因进行lasso回归分析,最终获得与预后相关的14个候选基因。将14个候选基因纳入双向逐步多因素Cox回归分析中,得到包含9个TME相关基因的ACC预后模型:RS=GREB1×0.223 6+POU4F1×0.671 7+HIC1×0.167 5+HOXC9×0.211 3+CACNB2×0.156 0+RAB27B×0.956 5+ZIC2×0.582 7+C3×(-0.003 1)+ CYP2D6×0.819 3(表1)。RS中位值为0.617 4分,依此将79例ACC患者分为RS低评分组(RS<0.617 4分,40例)和RS高评分组(RS≥0.617 4分,39例)。Kaplan-Meier生存曲线显示,RS高评分组患者总生存不良(P<0.05)(图3A)。绘制RS预测1、3、5年患者总生存的ROC曲线,曲线下面积(AUC)分别为0.876、0.919、0.917(图3B),提示该模型对生存结果的预测准确性较高。
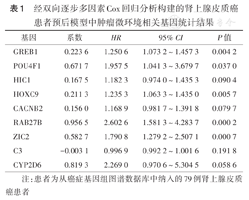
经双向逐步多因素Cox回归分析构建的肾上腺皮质癌患者预后模型中肿瘤微环境相关基因统计结果
经双向逐步多因素Cox回归分析构建的肾上腺皮质癌患者预后模型中肿瘤微环境相关基因统计结果
| 基因 | 系数 | HR | 95% CI | P值 |
|---|---|---|---|---|
| GREB1 | 0.223 6 | 1.250 6 | 1.073 2~1.457 3 | 0.004 2 |
| POU4F1 | 0.671 7 | 1.957 5 | 1.041 3~3.679 7 | 0.037 0 |
| HIC1 | 0.167 5 | 1.182 3 | 0.974 0~1.435 3 | 0.090 4 |
| HOXC9 | 0.211 3 | 1.235 3 | 1.063 3~1.435 0 | 0.005 7 |
| CACNB2 | 0.156 0 | 1.168 9 | 0.981 7~1.391 8 | 0.079 7 |
| RAB27B | 0.956 5 | 2.602 6 | 1.581 3~4.283 7 | 0.000 2 |
| ZIC2 | 0.582 7 | 1.790 8 | 1.279 2~2.507 1 | 0.000 7 |
| C3 | -0.003 1 | 0.996 9 | 0.992 2~1.001 6 | 0.191 8 |
| CYP2D6 | 0.819 3 | 2.269 0 | 0.970 6~5.304 5 | 0.058 6 |
注:患者为从癌症基因组图谱数据库中纳入的79例肾上腺皮质癌患者
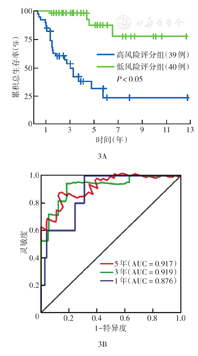
注:AUC为曲线下面积;患者为从癌症基因组图谱数据库中纳入的肾上腺皮质癌患者;依据风险评分中位值0.617 4分分为高风险评分(≥0.617 4分)组和低风险评分(<0.617 4分)组
从GEO数据库下载GSE33371和GSE19750芯片数据集中ACC患者资料,分别有23、22例,依据预测模型,对此45例患者生存进行分析。Kaplan-Meier生存曲线分析显示,建立的预后模型高、低RS组间总生存差异有统计学意义(P<0.05)(图4A)。绘制RS预测ACC患者1、3、5年总生存的ROC曲线,AUC分别为0.689、0.704、0.708(图4B),提示该模型对生存具有较好的预测性能。
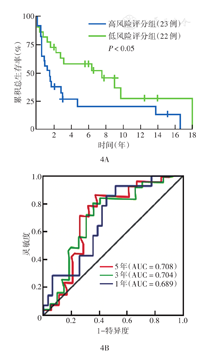
注:AUC为曲线下面积;患者为从GEO数据库下载GSE33371和GSE19750芯片数据集中纳入的肾上腺皮质癌患者;依据风险评分中位值0.617 4分分为高风险评分(≥0.617 4分)组和低风险评分(<0.617 4分)组
从TCGA数据库中提取到79例ACC患者的完整临床特征,其中年龄≤65岁71例,>65岁8例;男性50例,女性29例;Ⅰ期9例,Ⅱ期38例,Ⅲ期16例,Ⅳ期16例;T1期9例,T2期43例,T3期9例,T4期18例;M0期63例,M1期16例;N0期69例,N1期10例。单因素Cox回归分析显示,病理分期、肿瘤大小、远处转移情况和ACC预后模型RS是ACC患者总生存的危险因素(均P<0.05);将这些因素纳入多因素Cox回归分析,结果显示ACC预后模型RS是ACC患者预后的独立影响因素(HR=1.011,95% CI 1.005~1.016,P<0.01)。
近年来,随着对免疫治疗机制和靶向治疗机制的深入研究,TME受到越来越多的关注。Kitawaki等[7]首先对自主分泌激素的肾上腺皮质腺瘤的TME进行了组织学研究,证明在产生皮质醇的肾上腺腺瘤中,免疫细胞的浸润有特异性。Armignacco等[8]研究发现脂肪干细胞作为TME的关键成分,介导了ACC细胞的代谢,促进了癌细胞的侵袭;免疫治疗的阳性反应通常取决于肿瘤细胞之间的相互作用和TME的免疫调节。Wood等[9]阐述了自分泌、旁分泌信号间相互作用,细胞外基质重建以及肿瘤细胞与周围间质之间相互作用的机制,这些机制为非小细胞肺癌患者的靶向治疗提供了重要的理论基础。Petitprez等[10]研究显示,肿瘤细胞和TME成分之间的相互作用取决于原始器官、治疗类型和致癌过程。2013年Yoshihara等[5]首次提出了基于基因表达数据的免疫细胞和基质细胞浸润的ESTIMATE算法。这有助于阐明TME对肿瘤细胞作用的机制,也为基因组的深入研究提供了新方法。本研究的特点是免疫评分、基质评分和ESTIMATE评分均采用此算法来计算。结果显示,免疫评分降低与ACC临床分期升高相关(P=0.016)。进一步的Kaplan-Meier生存曲线分析表明,低免疫评分组总生存不佳(P<0.05)。而与其他临床病理特征的比较也表明构建的预后模型RS是ACC患者预后的独立危险因素(P<0.01)。与其他研究不同的是,本研究基于高质量数据集的转录谱确定了ACC TME中与免疫细胞和基质细胞浸润相关的特异性基因,而不是仅聚焦于TME中的免疫分子和肿瘤细胞。
目前本研究预后模型筛选出的9个TME相关基因中,部分基因在其他肿瘤中有过报道。Shin等[11]发现GREB1是一种具有进化保守结构域的蛋白质,可将ERα糖基化和稳定性与癌症联系起来。据报道,在类似卵巢性索肿瘤的子宫肌瘤患者中也检测到该基因[12,13]。Matsumoto等[14]报道GREB1是Wnt-catenin信号通路转导的靶分子,通过抑制TGFβ信号促进肝母细胞瘤的发展。POU4F1是POU域家族转录因子的成员,在调节癌症中具有关键作用。Wu等[15]通过实验验证POU4F1通过调节ERK1/2信号通路,可导致人表皮生长因子受体2(HER2)阳性乳腺癌曲妥珠单抗耐药。此外,POU4F1作为一种干细胞相关转录因子,在黑色素瘤细胞中高表达,有助于BRAF激活的恶性转化。Liu等[16]发现POU4F1通过激活MEK-ERK通路和上调MITF来促进黑色素瘤对BRAF抑制剂的耐药性。这些均表明POU4F1是癌症治疗中有前景的预后和治疗靶点。HIC1是位于17p13.3染色体的肿瘤抑制基因,是编码参与DNA损伤反应的转录抑制因子[17]。Dubuissez等[18]发现HIC1通过表观遗传修饰调节前列腺癌的进展。HOXC9在多种恶性肿瘤中高表达,Zhao等[19]在胃癌患者中观察到HOXC9过表达,并且与不良预后相关。Lv等[20]报道了膀胱癌细胞中miR-193a-3p或HOXC9水平的强制逆转极大地改变了DNA损伤反应和氧化应激途径的活性,miR-193a-3p通过靶向HOXC9基因来促进膀胱癌的多药耐药性。Cheng等[21]研究显示,RAB27B激活分泌的干细胞样肿瘤外泌体提供生物标志物miR-146a-5p,在结直肠癌中促进肿瘤发生并与免疫抑制TME相关,这与我们的研究结果相一致。ZIC2作为一种转录因子,可以与各种DNA和蛋白质相互作用。研究表明,ZIC2在多种癌症中发挥致癌基因作用[22]。Xu等[23]研究显示ZIC2在结肠癌组织中高表达,并与生存不良有关。他们的研究结果揭示了ZIC2在结肠癌中致癌活性的新多层次机制,表明ZIC2可作为结肠癌患者的潜在治疗靶点。Lu等[24]研究发现ZIC2通过PAK4促进肝细胞癌的肿瘤生长和转移。因此,我们认为GREB1、POU4F1、HOXC9、RAB27B、ZIC2可能是促进肿瘤发生和进展的微环境相关基因。
总之,我们构建的预后模型中TME相关基因与ACC等肿瘤的发生、发展关系密切,这些基因有作为ACC研究特异性分子标志物的潜质,也可能作为预后评估的指标,未来还可能作为研究发病机制和免疫治疗的新靶点。尽管我们系统地揭示了TME相关基因对于ACC患者预后评估的重要作用,但依旧存在一定的局限性:首先,预后模型的预测能力仍需大量多中心的临床证据支持,我们通过生物信息学方法在公共数据库的基础上进行了本研究,对其他人群的适用性还有待检验;其次,这9个与TME相关的关键基因还需要进一步研究,以更好地了解免疫浸润的调控机制,这可能会为全面研究TME与预后的潜在关联带来新的见解。但本模型仍能提供一定的预后评价指导,有助于制订个性化ACC治疗方案,使患者获益。
所有作者均声明不存在利益冲突

























