
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
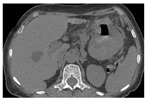
患者 女性,61岁,因上腹部不适7个月加重伴反复呕血、黑便2 d,于2014年10月16日来我院就诊,发病过程中无发热。查体:贫血貌,皮肤黏膜苍白,浅表淋巴结未触及肿大,腹平坦,未见肠型及蠕动波。血细胞分析(全血分类):红细胞计数3.38×1012/L、血红蛋白89.00 g/L、白细胞计数6.08×109/L、淋巴细胞计数1.03×109/L、淋巴细胞比率16.92 %。肝功能检查示:丙氨酸氨基转移酶4.40 U/L、天门冬氨酸氨基转移酶10.90 U/L、总蛋白64.40 g/L、清蛋白(A) 34.40 g/L、球蛋白(G)30.00 g/L、A/G 1.15、β2-微球蛋白(β2-MG) 4.59 mg/L。免疫球蛋白及骨髓穿刺未见异常。胃镜示:胃体下部至胃窦近幽门口见有结节状肿物,边界不清,胃窦腔狭窄、僵硬,肿物表面溃疡糜烂,见有红黑色血迹,散在溃疡,覆有灰白苔,组织硬,脆,易出血。CT检查结果示:胃体处胃壁明显增厚改变,胃腔狭窄;周围可见淋巴结影,脾脏增大,密度均匀(图1)。无传染病史,行下部胃大部+网膜切除术。病理检查结果:胃大弯长24.5 cm,小弯长6.0 cm,上切缘直径6.0 cm,下切缘直径2.0 cm,胃体大弯侧见肿物10 cm×7 cm×6 cm,肿物向胃腔内结节状生长,周边呈溃疡状,切面灰白,质嫩,浸润胃壁肌层,大网膜组织与胃浆膜层广泛粘连,胃周脂肪组织见肿大淋巴结9枚,直径0.2~ 2.0 cm。显微镜下观察:大淋巴细胞样相似于免疫母细胞的瘤细胞弥漫浸润,分布于胃壁,最深达深肌层,组织伴大面积坏死。细胞形态单一,胞质丰富,细胞核大而圆,染色质粗,核仁小,有的不明显,见浆样分化的细胞,核分裂象易见,可见凋亡小体及吞噬可染小体的巨噬细胞(图2)。免疫组织化学示:MUM-1+(图3)、CD20-、CD3-、bcl-2-、CD8-、CD30-、CD38-、CD138-、CD10-、bcl-6-、CK-、Ki-67 60 %+、CD21-。原位杂交EBER结果阳性。病理诊断结果:胃体大弯非霍奇金浆母细胞淋巴瘤(PBL),胃周淋巴结转移(2/9)。
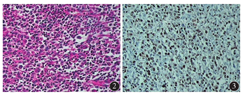
PBL是一类以大肿瘤细胞弥漫性增生的疾病,大部分肿瘤类似于B免疫母细胞,但是所有肿瘤细胞均表达浆细胞免疫表型。最早于口腔中发现这类肿瘤,结外部位更为常见。PBL是一种少见的淋巴瘤,好发于人类免疫缺陷病毒(HIV)阳性患者,男性更常见[1]。其他免疫缺陷患者和老年人也可发生,中位年龄约为50岁,年龄跨度较大,主要发生于成年人。极少数病例可见于伴有免疫缺陷的儿童[2]。好发于口腔黏膜,其他结外部位也可发生,包括鼻窦、眼眶、皮肤、骨、软组织和胃肠道。临床除有免疫抑制病症以外,局部包块为其特点。少数患者先存在浆细胞骨髓瘤,后转化为PBL。PBL的病因和发病机制仍未阐明。目前认为PBL起源于生发中心后的终末分化期的活化B细胞,可能处于免疫母细胞发育转化成浆细胞阶段,这些细胞已经历过体细胞、高频突变和免疫球蛋白(Ig)的类别转换。在这过程中,细胞内分子信号通路和染色体异常可导致细胞恶变[3]。
病理组织学特点如本病例显微镜下所见。免疫表型特征:PBL具有终末分化B细胞免疫表型,一般不表达或弱表达成熟B细胞的标志物CD20和Pax,而常表达浆细胞的标志物CD138、CD38、Vs38c、IRF4/MUM-1; 50 %~85%的患者CD79a阳性;50 %~75%的患者浆细胞Ig阳性,主要是IgG、Igκ或Igλ;肿瘤细胞也常表达CD30和上皮膜抗原(EMA)。发生于口腔的PBL CD56通常阴性,但常伴有浆细胞样型者CD56可以阳性。Ki-67指数常高于90%。60%~75%的病例EBER阳性,但很少表达潜伏膜蛋白1(LMP1)。PBL口腔黏膜型几乎100 %EB病毒(EBV)阳性,HHV8阳性也有报道。PBL患者特别是EBV阳性患者,易发生克隆性的IgH链基因以及myc基因的重排[4]。本例CD38、CD138均阴性,MUM-1阳性。
PBL在形态学上需要与浆细胞骨髓瘤、弥漫性大B细胞淋巴瘤、间变性大细胞淋巴瘤、Burkitt淋巴瘤、淋巴母细胞淋巴瘤相鉴别。本例发生于胃,还需要与低分化或未分化癌相鉴别。
PBL恶性程度高,预后极差。化疗药敏感性低,没有标准化疗方案,由于其临床及病理特点不够典型,所以要综合临床特征、形态学特点、免疫组织化学分型等多方面因素进行分析。大部分患者就诊时已处于临床Ⅲ或Ⅳ期,诊断后1.5年内死亡[5]。

























