
探讨以急变期为首发的慢性粒细胞白血病(CML)患者的临床和实验室特征。
分析2015年1月至2016年11月郑州大学附属洛阳中心医院收治的3例以急变期为首发的CML患者的临床和实验室特征及诊疗经过,患者首发症状均类似急性白血病。
3例患者主要表现均为脾大、外周血白细胞计数明显升高、骨髓中原始细胞大量增殖、骨髓中期相细胞费城染色体阳性(Ph+),荧光原位杂交分析分叶粒细胞可检测到bcr-abl1融合信号,bcr-abl1融合基因p210阳性,缺乏典型的CML慢性期病史。
以急变期为首发的CML患者由于骨髓中存在大量原始细胞,且临床表现类似于急性白血病,从而在初诊时易被误诊为Ph+急性白血病,临床表现、遗传学及基因组学有助于鉴别诊断。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
慢性粒细胞白血病(CML)是一种源于多能造血干细胞的恶性克隆性疾病,根据其自然病程可分为慢性期(CP)、加速期(AP)和急变期(BP)。多数病例起病缓慢,患者确诊时处于CP,可持续2~3年,随后进入AP和BP,而未经历前两个阶段,起病即进入BP,临床表现类似急性白血病的CML患者十分罕见。本文报道我科收治的3例以急性白血病为首发的CML患者,对其临床和实验室特征进行分析。
3例患者均为2015年1月至2016年11月我院门诊或住院患者。其中男性2例,女性1例,中位年龄52岁(46~55岁)。3例患者入院时均表现为脾大、外周血白细胞计数明显升高、骨髓原始细胞大量增生,根据2008年世界卫生组织(WHO)急性白血病诊断标准初步诊断3例患者分别为急性B淋巴细胞白血病(B-ALL)、急性髓系白血病(AML)-M2、急性双克隆(淋巴细胞-单核细胞)白血病,后根据患者临床表现、染色体核型、荧光原位杂交(FISH)、bcr-abl1融合基因等诊断患者为CML-BP。
对患者外周血涂片进行细胞形态学检查,对骨髓涂片进行常规细胞形态学及细胞化学检查。
使用BD FACSCantoⅡ型流式细胞仪和单克隆抗体(美国Becton Dickinson公司)对患者白血病细胞免疫表型进行分析。判断标准为胞质抗体≥10%为阳性,胞膜抗体≥20%为阳性。
取患者肝素抗凝骨髓5 ml,直接法或24 h短期培养法培养后,按常规方法制备染色体标本并采用G显带分析染色体核型,根据《人类细胞遗传学国际命名体制(ISCN)(2013)》进行异常核型描述。
bcr-abl1双色双融合易位探针购自美国Abbott Vysis公司,-20 ℃保存备用。按照说明书进行FISH操作,使用Olympus BX53荧光显微镜观察间期及中期细胞荧光杂交信号,有一红一绿两融合荧光信号的细胞为bcr-abl1融合基因阳性,每个样本至少分析400个间期细胞,计算阳性细胞比例。
取乙二胺四乙酸(EDTA)或肝素抗凝骨髓标本,使用TRIzol(美国Invitrogen公司)法抽提RNA并反转录为cDNA。3例患者均进行扩增43种白血病常见融合基因的多重巢式聚合酶链反应(PCR)检测并确定bcr-abl1融合基因类型和拷贝数;对例2、例3患者进行AML预后相关基因突变检测:FLT3/ITD、C-kit/D816、NPM1(exon12)和CEBPA;对3例患者均进行bcr-abl1酪氨酸激酶区突变检测。
1例行CDVLD方案诱导化疗、HyperCVAD B方案巩固化疗联合酪氨酸激酶抑制剂(TKI)治疗,并采用异基因造血干细胞移植(allo-HSCT)巩固治疗。1例行IA方案诱导、巩固化疗联合伊马替尼维持治疗。1例行改良CDVD方案诱导化疗联合伊马替尼维持治疗,但随后患者bcr-abl1酪氨酸激酶区发生F317L、D276G点突变,改用尼洛替尼维持治疗。
3例患者均具有完整的临床及血液学资料。初诊时中位脾脏大小分别为肋缘下8.0、3.5、2.0 cm;中位白细胞计数(WBC)为133.48×109/L[(96.07~174.68)×109/L],中位血小板计数(Plt)为121×109/L[(23~140)×109/L],中位血红蛋白(Hb)为51 g/L(45~119 g/L)。外周血分类显示例1嗜碱性粒细胞比例增高;例2嗜酸性粒细胞比例增高,原始细胞占0.30;例3原始细胞比例极高,占0.91(表1)。
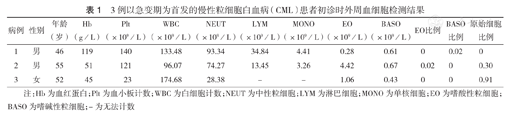
3例以急变期为首发的慢性粒细胞白血病(CML)患者初诊时外周血细胞检测结果
3例以急变期为首发的慢性粒细胞白血病(CML)患者初诊时外周血细胞检测结果
| 病例 | 性别 | 年龄(岁) | Hb(g/L) | Plt(×109/L) | WBC(×109/L) | NEUT(×109/L) | LYM(×109/L) | MONO(×109/L) | EO(×109/L) | BASO(×109/L) | EO比例 | BASO比例 | 原始细胞比例 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 男 | 46 | 119 | 140 | 133.48 | 93.34 | 34.84 | 4.41 | 0.28 | 0.61 | 0 | 0.02 | 0 |
| 2 | 男 | 55 | 51 | 121 | 96.07 | 74.27 | 13.45 | 3.26 | 4.42 | 0.67 | 0.02 | 0 | 0.30 |
| 3 | 女 | 52 | 45 | 23 | 174.68 | 28.38 | - | - | 1.06 | 0.43 | 0 | 0 | 0.91 |
注:Hb为血红蛋白;Plt为血小板计数;WBC为白细胞计数;NEUT为中性粒细胞;LYM为淋巴细胞;MONO为单核细胞;EO为嗜酸性粒细胞;BASO为嗜碱性粒细胞;-为无法计数
3例患者骨髓中原始细胞大量增殖,骨髓中期相细胞费城染色体阳性(Ph+),其中例1骨髓增生明显活跃,淋巴细胞恶性增殖,原始+幼稚淋巴细胞0.372;例2骨髓增生极度活跃,粒系0.712,原始粒细胞0.376;例3骨髓可见大小不等的两群白血病细胞,原始+幼稚淋巴细胞0.604,原始单核细胞0.328,粒系受抑制(0.024),嗜酸性粒细胞0.012。
按照2008年WHO分类,例1白血病细胞上的CD抗原表达归为B淋系表达;例2白血病细胞上的CD抗原表达归为髓系表达;例3患者具有两群白血病细胞,一群表达B淋系抗原,一群表达髓系抗原(偏单核方向分化)。
例1在治疗6个月随访结束时复查为正常核型。例2初诊时除具有t(9;22)(q34;q11)异常外,还附加额外的一条疑似10号染色体。例3初诊时除具有t(9;22)(q34;q11)异常外,还伴有dic(6;5)(q27;q31)和15号染色体丢失。3例患者FISH检测均可见bcr-abl融合信号,阳性率分别为78%、89%、95%。FISH分析发现,bcr-abl1融合信号并不局限于单个核细胞,分叶核细胞即成熟粒细胞也具有bcr-abl1融合信号(图1),考虑这3例患者可能为以急性白血病为首发的CML。3例患者的染色体核型结果及FISH结果见表2。
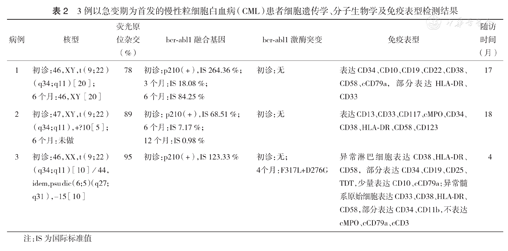
3例以急变期为首发的慢性粒细胞白血病(CML)患者细胞遗传学、分子生物学及免疫表型检测结果
3例以急变期为首发的慢性粒细胞白血病(CML)患者细胞遗传学、分子生物学及免疫表型检测结果
| 病例 | 核型 | 荧光原位杂交(%) | bcr⁃abl1融合基因 | bcr⁃abl1激酶突变 | 免疫表型 | 随访时间(月) |
|---|---|---|---|---|---|---|
| 1 | 初诊:46,XY,t(9;22)(q34;q11)[20];6个月:46,XY[20] | 78 | 初诊:p210(+),IS 264.36 %;3个月:IS 18.08 %;6个月:IS 84.25 % | 初诊:无 | 表达CD34、CD10、CD19、CD22、CD38、CD58、cCD79a,部分表达HLA⁃DR、CD33 | 17 |
| 2 | 初诊:47,XY,t(9;22)(q34;q11),+ ? 10[5];6个月:未做 | 89 | 初诊:p210(+),IS 68.51 %;6个月:IS 7.17 %;12个月:IS 0.98 % | 初诊:无 | 表达CD13、CD33、CD117、cMPO、CD34、CD38、HLA ⁃ DR、CD58、CD123 | 18 |
| 3 | 初诊:46,XX,t(9;22)(q34;q11)[10]/44,idem,psudic(6;5)(q27;q31),-15[10] | 95 | 初诊:p210(+),IS 123.33 % | 初诊:无;4个月:F317L+D276G | 异常淋巴细胞表达CD38、HLA ⁃ DR、CD58,部分表达CD34、CD19、CD25、TDT,少量表达CD10、cCD79a;异常髓系原始细胞表达CD33、CD38、HLA ⁃ DR、CD58,部分表达CD34、CD11b,不表达cMPO、cCD79a、cCD3 | 4 |
注:IS为国际标准值
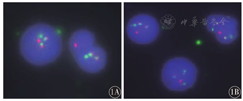
注:应用bcr-abl1双色双融合易位探针,正常信号为两红两绿(2R2G),阳性信号为一红一绿两融合(1R1G2F),在分叶核细胞即成熟粒细胞和原始细胞中都可以检测到bcr-abl1融合信号
3例患者均为p210融合基因阳性。初诊时3例患者均无bcr-abl1酪氨酸激酶区点突变,例3在改良CDVD方案诱导化疗联合伊马替尼维持治疗4个月后出现F317L+D276G点突变(表2)。例2和例3 FLT3/ITD、C-kit/D816、NPM1(exon12)和CEBPA突变均为阴性。
例1经诱导化疗联合伊马替尼治疗后获得完全缓解(CR),3个月时bcr-abl1国际标准值(IS)为18.08%,恢复至CP,随后行巩固化疗联合伊马替尼治疗,6个月时染色体核型为46,XY[20],bcr-abl1 IS为84.25%,患者bcr-abl1转录本下降不理想,行allo-HSCT。例2经诱导化疗联合伊马替尼治疗后获得CR,恢复至CP,随后行巩固化疗联合伊马替尼治疗,6个月时bcr-abl1 IS为7.17%,未行allo-HSCT,持续伊马替尼维持治疗,12个月时bcr-abl1 IS为0.98%,患者仍处于CP。例3经1个疗程诱导化疗获血液学缓解(WBC 8.87×109/L、Hb 83 g/L、Plt 112×109/L)后出院,院外口服伊马替尼3个月后复发,血常规示WBC 13.76×109/L、Hb 72 g/L、Plt 76×109/L,外周血分类示原始细胞0.51、嗜酸性粒细胞0.03,检出bcr-abl1酪氨酸激酶区F317L+D276G点突变,换用尼洛替尼维持治疗,至截稿时仍在随访中。
t(9;22)(q34;q11.2)染色体易位累及9号染色体长臂(9q34)上的原癌基因abl和22号染色体(22q11)上的原癌基因bcr,形成特征性Ph染色体和具有酪氨酸激酶活性的bcr-abl1融合蛋白[1,2,3]。Ph染色体见于几乎所有的CML、5%儿童ALL、15%~30%成年人ALL以及1%~2%急性髓系白血病(AML)患者[4,5]。
CML的自然病程通常包括2~3个临床分期,BP是CML的终末阶段,患者一旦进入BP,预后差。从CP向AP或BP演进是CML自然病程的一部分,大部分患者由CP经过AP,最终进入BP[6,7]。本研究报道的3例患者均脾大、白细胞计数明显升高,经外周血形态分析,骨髓细胞学形态分析及流式细胞术免疫分型后初步诊断3例患者分别为B-ALL、AML-M2、急性双克隆白血病,后根据细胞遗传学、分子生物学检测结果及患者的临床表现,考虑这3例患者为CML-BP,可能为以BP为首发的CML。根据已有报道,仅有10%的CML患者表现为首发急变,该部分患者由于骨髓中存在大量原始细胞且临床表现也类似于急性白血病,从而在初诊时易被误诊为急性白血病[6,7]。目前对于以BP为首发的CML与原发急性白血病的鉴别,大部分学者认为常规染色体核型分析是重要方法之一[8],而对于以BP为首发的CML和Ph+急性白血病的鉴别有待于进一步研究。
1975年Sasaki等[9]首次报道了2例Ph+ AML,1983年该研究小组对10例Ph+ AML的染色体核型进行分析,结果提示多数Ph+ AML患者的染色体核型中存在正常核型(占6%~65%)[10],绝大多数Ph+ CML患者的染色体核型中不存在正常核型[10,11]。本研究报道的3例患者中期相细胞均存在Ph染色体,与文献报道相符。Kantarjian等[12]对32例Ph+急性白血病患者bcr-abl1融合基因的研究结果提示,bcr-abl1 p210和p190的检测可能可用于鉴别CML-BP和初诊Ph+急性白血病,p210阳性可作为CML的特异性标志,而p190阳性则更易发生于初治Ph+急性白血病。本研究报道的3例患者均为p210阳性,因此考虑患者可能为CML急变而非Ph+急性白血病。Soupir等[13]对Ph+ AML的形态特征、免疫表型、染色体核型及bcr-abl1融合基因的多中心回顾性研究结果表明,与CML髓系急变相比,Ph+ AML患者较少发生脾大、外周血和骨髓中嗜碱性粒细胞较少、骨髓象中粒/红细胞比例较低。本文报道的例2和例3患者均具有明显的脾大、例2患者外周血嗜酸粒细胞升高、例3患者诱导化疗联合伊马替尼治疗复发后外周血嗜酸性细胞升高,与文献报道一致。2013年Konoplev等[14]从分子生物学水平上对9例Ph+ AML和5例CML-BP患者进行研究,结果提示约20% CML-BP患者具有bcr-abl1激酶突变但不发生NPM1突变,而Ph+ AML患者可发生NPM1突变而不发生bcr-abl1激酶突变。本研究报道的例2、例3患者初诊bcr-abl1激酶区突变检测及NPM1基因突变均为阴性,但例3患者在诱导化疗及伊马替尼联合治疗4个月后发生bcr-abl1激酶区突变。Nacheva等[15]对Ph+急性白血病进行的以阵列为基础的比较基因组杂交(aCGH)研究结果验证了Konoplev等[14]的研究结果,同时提示在基因组水平上Ph+ AML与Ph+ ALL和CML-BP截然不同。
2013年Choi等[16]报道了4例以急性白血病为首发的CML,该4例患者经过一系列临床和实验室检查后被初步考虑为Ph+急性白血病,但经过对成熟粒细胞进行FISH分析检测bcr-abl1融合信号,加以染色体核型分析及bcr-abl1融合基因检测后,诊断为CML-BP。本研究报道的3例患者均在成熟粒细胞中检出bcr-abl1融合信号。2016年Kamoda等[17]对20例无CML病史的Ph+ ALL患者进行FISH分析发现,9例在成熟粒细胞中可检出bcr-abl1融合信号的患者与成熟粒细胞bcr-abl1融合信号阴性患者相比,bcr-abl1融合基因多为p210阳性,且有更高的WBC和更好的无病生存和总生存,提示该部分ALL患者可能为CML急淋变。本研究报道与文献相符。因CML为多能造血干细胞异常,在所有造血系细胞中均可发现t(9;22)易位,且在CML中白血病细胞主要向中性粒细胞分化[6],因而在成熟中性粒细胞中检测到bcr-abl1融合基因有望作为CML-BP的诊断特征。
综上,我们可依据患者的临床特点、形态学特征、细胞遗传学特征(染色体核型、FISH检测成熟中性粒细胞bcr-abl1融合基因)、分子生物学特征(bcr-abl1融合基因、bcr-abl1激酶、NPM1突变检测)及基因组水平的检测来对以BP为首发的CML和Ph+急性白血病进行鉴别。
无

























