
探讨急性早幼粒细胞白血病(APL)附加染色体异常及t(15;17)的临床意义。
回顾性分析2007年1月至2014年6月青岛大学附属医院收治的90例初治APL患者的临床特点及染色体资料。根据染色体核型的不同分为附加仅数目改变组(16例)、附加仅结构异常组(14例)、附加数目改变和结构异常组(4例)以及典型染色体组(56例)。按照是否含有t(15;17)分为含有t(15;17)组(82例)和不含有t(15;17)组(8例)。分析比较各组近期疗效及生存情况。
附加仅数目改变组、附加仅结构异常组、附加数目改变和结构异常组以及典型染色体组的完全缓解率分别为56.3%(9/16)、100.0%(14/14)、25.0%(1/4)和82.1%(46/56),早期的死亡率分别为25.0%(4/16)、0(0/14)、75.0%(3/4)和8.9%(5/56),其中附加数目改变和结构异常组完全缓解率低,早期死亡率高,与其他组比较,差异均有统计学意义(均P<0.05)。含有t(15;17)组和不含有t(15;17)组完全缓解率分别为80.5%(66/82)和50.0%(4/8),差异无统计学意义(P=0.070)。
染色体核型同时存在附加数目和结构改变的APL患者完全缓解率低,早期死亡率高,预后不良。含有t(15;17)的患者完全缓解率较高。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
急性早幼粒细胞白血病(APL)类型特殊,存在特殊的细胞遗传学标志,最典型的改变为第15号和第17号染色体长臂发生平衡易位,形成特异染色体改变和融合基因产物PML-RARα。随着靶向治疗药物维甲酸的应用,APL患者可获得90%以上的完全缓解率。但也有文献报道t(5;17)(q32;q21)、t(11;17)(q13;q21)和t(11;17)(q23;q21)这3种少见的变异型易位,分别形成RARα基因的伙伴基因核仁磷酸蛋白(NPM)、核有丝分裂器(NuMA)和早幼粒细胞白血病锌指(PLZF)基因[1],携带该类基因的患者对维甲酸治疗不敏感,但是可通过三氧化二砷诱导缓解。近年来国内外陆续报道了除典型t(15;17)(q22;q21)改变外的附加染色体异常,虽然有研究提出ider(17q)提示预后不良,但附加染色体异常的意义始终存在争议。本研究探讨了APL附加染色体变异及t(15;17)的临床意义。
收集2007年1月至2014年6月我科初诊的APL患者90例,其中男性46例,女性44例,中位年龄41岁(14~78岁)。
采用骨髓细胞24 h短期培养法,按常规制备染色体。应用G显带技术进行核型分析,核型异常按人类细胞遗传学命名国际体制(ISCN)1995加以描述。根据染色体核型的不同分为附加仅数目改变组、附加仅结构异常组、附加数目改变和结构异常组、典型染色体组。按照是否含有典型染色体改变t(15;17)分为含有t(15;17)组和不含有t(15;17)组。
APL及弥散性血管内凝血(DIC)的诊断均符合文献[2]标准。给予全反式维甲酸每天25 mg/m2+三氧化二砷10 mg/d±DA(柔红霉素+阿糖胞苷)方案、IDA(去甲氧柔红霉素+阿糖胞苷)方案、MA(米托蒽醌+阿糖胞苷)方案诱导缓解治疗。根据病情变化给予血制品支持治疗,并针对性预防肿瘤溶解综合征和维甲酸综合征的发生。经骨髓形态学证实达到完全缓解后给予巩固化疗,三氧化二砷维持,或单月、每周使用甲氨蝶呤和每天服用巯嘌呤,持续3年。如发生中枢神经系统白血病,给予鞘内注射化疗药物与放疗联合。
采用SPSS 17.0软件对数据进行统计学分析,计数资料的比较采用Fisher确切概率法,以P<0.05为差异有统计学意义。
90例初治APL患者中,典型染色体改变56例,附加染色体变异核型34例(37.8%),包括核型仅发生附加数目改变16例(常染色体11例,性染色体3例,标记染色体2例),仅附加结构异常14例,附加结构与数目同时改变4例。常染色体数目改变以+8为主,6例为t(15;17)伴有+8,1例为t(15;17)同时伴有+8、+13、+21、+i(17q-),其他还有亚克隆+8及超二倍体+13、+14、+21、+22。染色体结构异常以缺失、等臂染色体、亚克隆及易位为主。染色体缺失分别发生在8q22、del(9)(q11q22)、del(7)(q22q34),末端缺失同时伴t(15;17),中间缺失无t(15;17)改变;等臂染色体多发生在17q,且同时伴t(15;17);易位共有3例,分别为t(14;17)(q32;q21)、t(15;17)合并t(5;12)或t(17;18)。肿瘤细胞基本的克隆干系为t(15;17)或t(15;17)伴有add(8)(p11)。附加结构与数目同时改变者均伴t(15;17),包括:常染色体数目缺失[-14、-15、-20、-21、-22、-19、del(16)(q11)]、性染色体改变为-X;结构改变为有1~4条标记染色体加入,第1、17号染色体多处缺失,第21、22号染色体缺失导致的衍生染色体的出现。第17号衍生染色体的出现同时伴有7q22缺失、13p11上未知来源片段的加入、标记染色体的加入。
附加仅数目改变组、附加仅结构异常组、附加数目改变和结构异常组以及典型染色体组患者的性别、肿瘤浸润、白细胞计数、血小板计数及DIC情况比较,差异均无统计学意义(均P>0.05)(表1)。
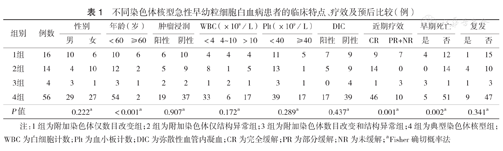
不同染色体核型急性早幼粒细胞白血病患者的临床特点、疗效及预后比较(例)
不同染色体核型急性早幼粒细胞白血病患者的临床特点、疗效及预后比较(例)
| 组别 | 例数 | 性别 | 年龄(岁) | 肿瘤浸润 | WBC(×109/L) | Plt(×109/L) | DIC | 近期疗效 | 早期死亡 | 复发 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 男 | 女 | <60 | ≥60 | 阳性 | 阴性 | <4 | 4~10 | >10 | <40 | ≥40 | 阳性 | 阴性 | CR | PR+NR | 是 | 否 | 是 | 否 | ||
| 1组 | 16 | 10 | 6 | 10 | 6 | 6 | 10 | 4 | 4 | 4 | 11 | 5 | 7 | 9 | 9 | 7 | 4 | 12 | 1 | 15 |
| 2组 | 14 | 4 | 10 | 12 | 2 | 5 | 9 | 8 | 1 | 5 | 13 | 1 | 5 | 9 | 14 | 0 | 0 | 14 | 4 | 10 |
| 3组 | 4 | 3 | 1 | 3 | 1 | 2 | 2 | 1 | 2 | 1 | 3 | 1 | 0 | 4 | 1 | 3 | 3 | 1 | 1 | 3 |
| 4组 | 56 | 29 | 27 | 54 | 2 | 19 | 37 | 33 | 6 | 17 | 39 | 17 | 17 | 39 | 46 | 10 | 5 | 51 | 9 | 47 |
| P值 | 0.222a | <0.001a | 0.907a | 0.172a | 0.289a | 0.437a | 0.001a | 0.002a | 0.341a | |||||||||||
注:1组为附加染色体仅数目改变组;2组为附加染色体仅结构异常组;3组为附加染色体数目改变和结构异常组;4组为典型染色体核型组;WBC为白细胞计数;Plt为血小板计数;DIC为弥散性血管内凝血;CR为完全缓解;PR为部分缓解;NR为未缓解;aFisher确切概率法
附加仅数目改变组16例患者中,6例(37.5%)发病年龄≥60岁;9例(56.3%)经标准诱导治疗1个疗程达到完全缓解;1例在完全缓解19个月后复发,4例(25.0%)早期死亡。附加仅结构异常组14例患者中,2例(14.3%)发病年龄≥60岁;14例(100.0%)均经标准诱导治疗1个疗程达到完全缓解;4例复发,复发时间为0.5~4年,无早期死亡者。附加数目改变和结构异常组4例患者中,1例(25.0%)发病年龄≥60岁;1例(25.0%)经标准诱导治疗1个疗程达到完全缓解;1例在5年后复发,3例(75.0%)早期死亡。典型染色体组56例患者中,2例(3.6%)发病年龄≥60岁;46例(82.1%)经1个疗程标准诱导化疗达到完全缓解;9例复发,复发时间4~60个月,5例(8.9%)早期死亡。附加数目改变和结构异常组完全缓解率低,早期死亡率高,与其他组比较,差异均有统计学意义(均P<0.05);附加仅数目改变组发病年龄≥60岁患者比例高于其他三组,差异有统计学意义(P<0.05)(表1)。
含有t(15;17)患者82例,不含有t(15;17)患者8例,两组患者的性别、年龄、肿瘤浸润、白细胞计数、血小板计数及DIC情况比较,差异均无统计学意义(均P>0.05)(表2)。含有t(15;17)患者中,66例(80.5%)经1个疗程标准诱导化疗达到完全缓解;10例复发,复发时间4~60个月,9例(11.0%)早期死亡。不含有t(15;17)患者中,4例(50.0%)经1个疗程标准诱导化疗达到完全缓解;2例复发,复发时间分别在完全缓解后第8、19个月,2例(25.0%)早期死亡。含有t(15;17)组和不含有t(15;17)组完全缓解率比较,差异无统计学意义(P=0.070)。
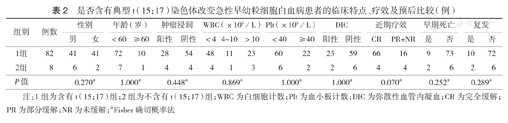
是否含有典型t(15;17)染色体改变急性早幼粒细胞白血病患者的临床特点、疗效及预后比较(例)
是否含有典型t(15;17)染色体改变急性早幼粒细胞白血病患者的临床特点、疗效及预后比较(例)
| 组别 | 例数 | 性别 | 年龄(岁) | 肿瘤浸润 | WBC(×109/L) | Plt(×109/L) | DIC | 近期疗效 | 早期死亡 | 复发 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 男 | 女 | <60 | ≥60 | 阳性 | 阴性 | <4 | 4~10 | >10 | <40 | ≥40 | 阳性 | 阴性 | CR | PR+NR | 是 | 否 | 是 | 否 | ||
| 1组 | 82 | 41 | 41 | 72 | 10 | 28 | 54 | 48 | 11 | 23 | 60 | 22 | 23 | 59 | 66 | 16 | 9 | 73 | 10 | 72 |
| 2组 | 8 | 6 | 2 | 7 | 1 | 4 | 4 | 4 | 1 | 3 | 6 | 2 | 2 | 6 | 4 | 4 | 2 | 6 | 2 | 6 |
| P值 | 0.270a | 1.000a | 0.448a | 0.869a | 1.000a | 1.000a | 0.070a | 0.252a | 0.289a | |||||||||||
注:1组为含有t(15;17)组;2组为不含有t(15;17)组;WBC为白细胞计数;Plt为血小板计数;DIC为弥散性血管内凝血;CR为完全缓解;PR为部分缓解;NR为未缓解;aFisher确切概率法
APL除典型的t(15;17)改变外,还伴有少见的变异,这些患者往往治疗效果欠佳[1],近年来又陆续发现了新的附加染色体的改变[3,4],国外还报道了1例更为复杂的染色体易位核型,为46、XX,t(5;17;15)(q35;q21;q22)、add(7)(q32)[5]。这些附加染色体改变的意义目前尚无定论。
本研究中附加染色体变异的发生率为37.8%,与de Botton等[6]报道的25%~40%一致,但比路谨等[7]报道的18.4%高,可能与染色体检测方法不同有关。附加染色体数目改变以+8为主,对患者预后无影响,与既往多项相关临床报道一致[8,9,10]。本研究结果显示附加染色体数目和结构同时改变的核型患者完全缓解率低,早期死亡率高,提示二者同时存在为预后不良因素之一。但袁燕慧等[11]的研究显示,是否伴有附加染色体异常对预后无影响,与本研究结果不一致的原因可能是染色体分析方法不同及异常染色体分析中存在混杂因素。有研究[6,12,13]指出含有t(15;17)为APL患者预后良好因素,本研究中含有t(15;17)的APL患者完全缓解率为80.5%,但与不含有t(15;17)者的50.0%比较,差异无统计学意义(P>0.05),可能与样本量少有关。
有报道显示,伴少见融合基因STAT5b-RARα的APL预后差,明确诊断后根据病情及时调整治疗方案并联合异基因造血干细胞移植可能改善预后[14]。也有报道显示APL治疗后转变为M5型的病例临床较为罕见,转型后治疗效果不佳[15]。附加染色体数目和结构异常提示预后不良。临床医生需重视对患者病情的综合评估,进行诱导治疗过程中更要注意根据患者年龄进行化疗方案的选择和对症处理,以提高完全缓解率,减少不良反应和早期死亡发生。
所有作者均声明不存在利益冲突

























