
探讨PML-RARα不同亚型的急性早幼粒细胞白血病(APL)患者的临床特征及预后。
收集福建医科大学附属协和医院2013年2月至2016年7月收治的78例初诊APL患者的临床资料,分析PML-RARα不同亚型患者的临床特征及预后。
78例患者中,女性32例,男性46例,中位年龄40岁(13~68岁)。最常见的PML-RARα融合基因为L型(48.7%,38/78),其次为S型(46.2%,36/78)和V型(5.1%,4/78)。S型患者中白细胞计数>10×109/L(高危)者最多见(61.1%,22/36),与V型、L型比较,差异有统计学意义(χ2=7.683,P<0.05)。78例中,合并CD34阳性8例(10.2%),合并FLT3-内部串联重复(ITD)突变17例(21.8%),合并DNMT3A突变12例(15.4%),附加染色体异常9例(11.5%);3种不同亚型间CD34阳性、FLT3-ITD、DNMT3A以及附加染色体异常发生率差异无统计学意义(P>0.05)。S型患者治疗期间发生维甲酸综合征(RAS)最多见(21/36),L型及V型较少发生(χ2=7.633,P<0.05)。PML-RARα不同亚型患者间完全缓解率以及无病生存率差异均无统计学意义(P>0.05)。
PML-RARα不同亚型APL患者临床特点不尽相同,L型最常见,V型、S型中高危者多见,S型治疗期间合并RAS多见,不同亚型对完全缓解率及无病生存率无影响。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
急性早幼粒细胞白血病(APL)是急性白血病的一种特殊亚型,其特点为染色体t(15;17),第15号染色体上的PML基因和第17号染色体上的RARα基因融合后形成PML-RARα融合基因,抑制早幼粒细胞的分化,从而形成以异常早幼粒细胞增多为主的白血病[1,2]。在PML-RARα融合基因中,RARα基因长度为15~20 kb,其断裂位点为第2内含子,而PML基因的断裂部位集中在3个不同区域,分别是内含子6、外显子6和内含子3,分别与RARα基因融合形成L型(BCR1,约55%)、V型(BCR2,约5%)和S型(BCR3,约40%)3种APL亚型[3,4]。国内外关于PML-RARα融合基因3种亚型的研究结果不一[5,6],且国内相关研究病例数偏少[7]。本研究旨在进一步探讨PML-RARα融合基因3种亚型与APL的临床特征和预后的关系。
2013年2月至2016年7月我科收治的APL患者78例,其中男性46例,女性32例,年龄13~68岁,中位年龄40岁。其中L型38例(48.7%),V型4例(5.1%),S型36例(46.2%)。全部病例均经细胞形态学、免疫分型、细胞遗传学以及分子生物学检查,符合APL诊断标准[8]。初诊时白细胞计数(WBC)(0.5~117.3)×109/L,血红蛋白(Hb)43~129 g/L,血小板计数(Plt)(6~190)×109/L,纤维蛋白原(FIB)0.5~4.7 g/L,乳酸脱氢酶(LDH)140~1 032 U/L。根据Sanz评分进行危险度分层[9],初诊WBC≤10×109/L且Plt>40×109/L为低危组(22例),WBC≤10×109/L且Plt≤40×109/L为中危组(20例),WBC>10×109/L为高危组(36例)。
诱导治疗方案为全反式维甲酸(ATRA)(每天20 mg/m2)及亚砷酸(每天0.16 mg/kg,最大剂量10 mg/d),WBC≥10×109/L时联合去甲氧柔红霉素(每天8 mg/m2)或柔红霉素(每天45 mg/m2)化疗。诱导治疗期间根据临床表现、血常规及凝血功能检查,给予红细胞、血小板及血浆输注等对症支持治疗;诱导治疗期间若出现发热、体质量增加、头痛、肌肉骨骼疼痛、呼吸窘迫、胸腔积液、肺间质浸润、水肿等维甲酸综合征(RAS)的表现时给予糖皮质激素治疗。
对所有患者均进行血常规、骨髓常规、染色体、融合基因以及预后相关基因突变检测。采用实时荧光定量聚合酶链反应(RT-PCR)检测PML-RARα融合基因,分析比较L型、V型和S型三组间的临床特点、治疗反应以及预后。疗效判定参考国际协作组AML疗效判定标准[10]。
应用SPSS 23.0统计软件。计量资料为非正态分布,以中位数表示,组间比较采用秩和检验;计数资料组间比较采用χ2检验;生存分析采用Kaplan-Meier法,并进行log-rank检验。以P<0.05为差异有统计学意义。
PML-RARα不同亚型患者血常规、凝血功能、LDH比较见表1。结果显示,不同亚型患者WBC、凝血酶原时间差异均有统计学意义(均P<0.05)。
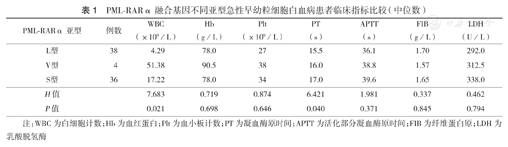
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者临床指标比较(中位数)
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者临床指标比较(中位数)
| PML-RARα亚型 | 例数 | WBC(×109/L) | Hb(g/L) | Plt(×109/L) | PT(s) | APTT(s) | FIB(g/L) | LDH(U/L) |
|---|---|---|---|---|---|---|---|---|
| L型 | 38 | 4.29 | 78.0 | 27 | 15.5 | 36.1 | 1.70 | 292.0 |
| V型 | 4 | 51.38 | 90.5 | 38 | 16.0 | 38.8 | 1.57 | 312.5 |
| S型 | 36 | 17.22 | 78.0 | 34 | 17.0 | 39.6 | 1.65 | 338.0 |
| H值 | 7.683 | 0.719 | 0.874 | 6.421 | 1.981 | 0.337 | 0.462 | |
| P值 | 0.021 | 0.698 | 0.646 | 0.040 | 0.371 | 0.845 | 0.794 |
注:WBC为白细胞计数;Hb为血红蛋白;Plt为血小板计数;PT为凝血酶原时间;APTT为活化部分凝血酶原时间;FIB为纤维蛋白原;LDH为乳酸脱氢酶
78例APL患者均进行免疫表型、基因突变以及附加染色体检测,其中,8例(10.2%)合并CD34阳性,17例(21.8%)合并FLT3-内部串联重复(ITD)突变,12例(15.4%)合并DNMT3A突变,9例(11.5%)检测到附加染色体异常[包括+8、add(8)、add(14)、inc(10)、add(4)](表2)。
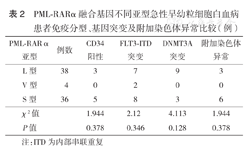
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者免疫分型、基因突变及附加染色体异常比较(例)
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者免疫分型、基因突变及附加染色体异常比较(例)
| PML-RARα亚型 | 例数 | CD34阳性 | FLT3-ITD突变 | DNMT3A突变 | 附加染色体异常 |
|---|---|---|---|---|---|
| L型 | 38 | 3 | 7 | 9 | 3 |
| V型 | 4 | 0 | 2 | 0 | 0 |
| S型 | 36 | 5 | 8 | 3 | 6 |
| χ2值 | 1.944 | 2.12 | 4.113 | 1.944 | |
| P值 | 0.378 | 0.346 | 0.128 | 0.378 |
注:ITD为内部串联重复
PML-RARα不同亚型患者APL危险度分层比较:L型中,低中危多见(68.4%,26/38),S型中,高危多见(61.1%,22/36),差异有统计学意义(P=0.039)(表3)。
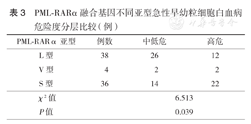
PML-RARα融合基因不同亚型急性早幼粒细胞白血病危险度分层比较(例)
PML-RARα融合基因不同亚型急性早幼粒细胞白血病危险度分层比较(例)
| PML-RARα亚型 | 例数 | 中低危 | 高危 |
|---|---|---|---|
| L型 | 38 | 26 | 12 |
| V型 | 4 | 2 | 2 |
| S型 | 36 | 14 | 22 |
| χ2值 | 6.513 | ||
| P值 | 0.039 | ||
78例患者接受诱导治疗后,除7例因颅内出血早期死亡外,其余均获得完全缓解(CR),L型、V型、S型患者诱导治疗的CR率分别为89.5%、75.0%、94.4%,差异无统计学意义(P>0.05)。诱导治疗期间34例(43.6%)出现RAS,不同亚型间发生率差异有统计学意义(P<0.05)(表4)。
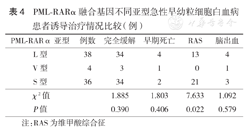
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者诱导治疗情况比较(例)
PML-RARα融合基因不同亚型急性早幼粒细胞白血病患者诱导治疗情况比较(例)
| PML-RARα亚型 | 例数 | 完全缓解 | 早期死亡 | RAS | 脑出血 |
|---|---|---|---|---|---|
| L型 | 38 | 34 | 4 | 13 | 4 |
| V型 | 4 | 3 | 1 | 0 | 1 |
| S型 | 36 | 34 | 2 | 21 | 3 |
| χ2值 | 1.885 | 1.803 | 7.633 | 1.092 | |
| P值 | 0.390 | 0.406 | 0.022 | 0.579 |
注:RAS为维甲酸综合征
从初诊到随访截止(2018年7月),中位随访时间36个月,3年总生存(OS)率为100%(除外7例因诱导治疗颅内出血而早期死亡的患者);L型、V型、S型的预期中位无病生存(DFS)时间分别为55、41、54个月,3种亚型的中位DFS时间差异无统计学意义(P=0.655)。
APL是一种特殊类型的急性髓系白血病,伴有特征性核型改变和基因融合,即t(15;17)(q22;q21),形成PML-RARα融合基因,其蛋白产物导致细胞分化阻滞和凋亡不足,是APL发生的主要分子机制[11]。患者常合并凝血功能异常,易发生严重出血。PML-RARα阳性的APL发病人群以青中年为主,平均发病年龄44岁[12],本研究中患者发病的中位年龄为40岁,与报道相符。
PML基因断裂点不同,产生了3种不同类型的PML-RARα融合基因。本研究主要针对PML-RARα阳性APL不同亚型的临床特点、治疗反应以及预后等方面进行研究。有研究认为不同种族患者间3种亚型的发生率不同,国外的一项多中心临床研究数据显示拉丁美洲APL患者中L型、V型、S型发生率分别为78%、11%、11%,非拉丁美洲则为52%、5%、43%[13],而本研究中L型、V型、S型的发生率分别为48.7%、5.1%、46.2%,与非拉丁美洲患者更接近,与国内报道一致。因此目前认为APL患者中PML基因断裂的部位与种族遗传或者环境因素有关,不同的致病因素导致APL基因断裂点的不同[1]。因此分析APL患者的亚型分布及特点很有临床意义。本研究分析了78例患者的资料,发现PML-RARα不同亚型的APL患者临床特征存在一定差异。
Jurcic等[14]通过对63例APL患者的PML-RARα亚型进行回顾性分析,认为S型APL患者DFS时间和OS时间均较L型APL患者缩短,预后不良,可作为独立的预后指标。本研究结果表明,明确诊断的78例APL患者中,诊断时S型中WBC>10.0×109/L者占61.1%(22/36),L型占31.6%(12/38),S型、V型的白细胞总数明显高于L型,差异有统计学意义。根据危险度分层,初诊WBC>10×109/L属于高危组,而两组诱导CR率及中位DFS时间比较差异均无统计学意义。因此PML-RARα亚型可作为APL的独立的预后因素。
本研究中8例(10.3%)患者表达CD34,与文献[15]报道的16.28%的APL可见CD34阳性表达接近。8例CD34阳性的APL患者中有1例因脑出血早期死亡,与CD34阴性患者相比,差异无统计学意义。但部分报道显示CD34阳性的APL患者预后不良[16],且ATRA对CD34阳性的APL患者治疗效果差,早期死亡率及复发率高,还需进一步研究证实,考虑可能与随访时间较短有关。
本研究结果显示PML-RARα阳性APL中FLT3-ITD突变率为21.8%,与文献[17]报道的12%~38%一致;国外报道S型FLT3-ITD突变率较L型和V型高,认为合并FLT3-ITD突变与疾病不良预后有关[18],但本研究3种亚型预后差异无统计学意义,这种差异可能与种族差异有关,不排除样本量较少引起的误差。本研究中9例(11.5%)伴附加染色体异常,但并未发现附加染色体异常与治疗反应及预后相关。
APL患者的治疗缓解率高,DFS时间长,本研究中78例患者根据危险分层分别采用不同的方案进行诱导治疗,均获得CR。ATRA诱导治疗过程中43.5% APL患者出现RAS,与国内报道的46.4%相一致[19]。RAS是APL诱导治疗中常见的并发症,也是影响APL患者预后的一个重要因素,危险度越高则发生率越高。本研究36例S型APL患者中21例发生RAS,L型及V型少见,不同亚型比较差异有统计学意义,这个结果与其根据危险度分层时S型中高危者多见相符。
APL患者的预后可能与WBC、PML-RARα融合基因、达CR时间、CD34、HLA-DR免疫表型、不良预后基因突变以及巩固维持治疗方案等相关,虽本组患者中S型、V型的患者白细胞高于L型患者,但仍有其他影响因素对患者的预后造成影响,可能是导致组间CR率和DFS差异无统计学意义的原因之一。而且近年来对APL诊断、治疗、病情监测及预后判断等方面均取得了长足进展,高危APL的巩固方案强,医患宣教工作到位,医生治疗规范,患者治疗配合度高,也是本组患者高危组与中低危组CR率及DFS无差异的一个重要原因。
总之,PML-RARα不同亚型的APL患者临床特征并不完全一致,其中L型最常见,V型及S型患者初诊时WBC高,其中S型患者高危组最多见,且在诱导分化治疗中RAS的发生率最高。PML-RARα不同亚型不影响APL患者的诱导CR率以及DFS。对PML-RARα不同亚型APL患者临床特征的研究,有助于在现有危险度分层的基础上进一步评估患者的预后,从而指导个体化治疗。
所有作者均声明不存在利益冲突

























