
生物类似药具有与参照药相似的质量、疗效和安全性,同时具有价格优势,对实现我国居民用药的更高可及性具有非常重要的意义。为加深对生物类似药这一新兴概念的正确认识,规范我国生物类似药的临床用药,共识专家组参考国内外相关循证医学证据,结合临床用药体会,经充分讨论沟通,达成以下共识:(1)生物类似药与参照药疗效等同、安全性相似,临床上可以替代使用;(2)根据"适应证外推"原则,生物类似药可获得参照药具有相同作用机制的其他所有适应证,而且对外推适应证的疗效和安全性与参照药相似;(3)对于正在接受治疗的患者,临床医生可根据患者情况,决定是否由使用参照药转换成使用生物类似药。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
为规范我国生物类似药的注册上市,国家药品监督管理局(NMPA)于2015年颁布了《生物类似药研发与评价技术指导原则(试行)》。生物类似药凭借其与参照药在质量、疗效和安全性方面的相似性以及价格优势,对实现我国居民用药的更高可及性具有非常重要的意义。为加深对生物类似药这一新兴概念的正确认识,规范我国生物类似药的临床用药,共识专家组参考国内外相关循证医学证据,结合临床用药体会,经充分讨论沟通,达成本共识。
生物类似药是指在质量、安全性和有效性方面与已获准注册的参照药(通常指原研药)具有相似性的治疗用生物制品[1],其具有以下4个方面的重要特征[2]。(1)与参照药高度相似:由于生物药固有的自然属性,生物类似药在物理、化学和生物特性方面不可能与参照药完全一致,即使参照药的同一批次内或不同批次间,都可能存在细微的差异,因而生物类似药与参照药只能高度相似,允许存在不影响临床安全性和有效性的微小差异。(2)与参照药临床意义相同:生物类似药与参照药的临床表现无任何差异。(3)差异性被严格限制:只有在科学证据表明不影响安全性和有效性时,这种微小差异才被允许。生物类似药与参照药间允许的差异范围与参照药不同批次间允许的差异范围相同(图1)。(4)质量、安全性和有效性有严格标准:生物类似药是按照与其他药品同样严格的质量、安全和疗效标准批准的。我国生物类似药现行的审批监管流程与新生物制品一样,包括注册审批、生物制品的批签发和上市后监管三大流程[3]。
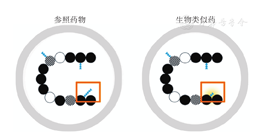
注:蛋白质氨基酸序列(圆形)一致,允许糖基化(蓝色小三角形)方面的差异(红框内黄色阴影)
随着越来越多的原研生物药专利到期,在临床需求、医保控费、商业效益等因素的共同推动下,当前全球生物类似药呈现出如火如荼的发展景象。检索Clarivate Analytics Cortellis数据库,截至2019年5月29日,全球正在研究的生物类似药有718个,包括上市的121个、临床研究阶段的155个、临床前研究阶段的386个;其中,单克隆抗体占全部生物类似药的52.4%。生物类似药的研发主要集中在肿瘤、免疫及血液系统疾病,这三大领域研发数量占56%。我国目前有27个生物类似药处于研发状态,其中约一半处于临床前研究阶段,有10个已获得临床研究批准。至2019年12月,我国共有4个生物类似药获批上市。
化学仿制药是与参照药具有相同活性成分、剂型、给药途径和治疗作用的药品。生物类似药之所以不称为仿制药,是因为生物药的自然变异性和复杂的生产工艺导致生物类似药与参照药只能高度相似。生物类似药与化学仿制药的具体区别见表1。

生物类似药与化学仿制药的区别
生物类似药与化学仿制药的区别
| 对比角度 | 生物类似药 | 化学仿制药 |
|---|---|---|
| 生产来源 | 生物来源,工艺更复杂 | 化学合成 |
| 分子结构 | 与参照药高度相似,通常是更大、更复杂的分子,需要多种技术鉴定 | 与参照药完全相同,大多数是小分子,容易鉴定 |
| 研究侧重点 | 基于生物相似性研究a | 基于生物等效性研究b |
| 药学研究 | 全面的药物质量数据和质量比对研究 | 全面的药物质量数据 |
| 临床研究 | 药代动力学生物等效性、安全性和有效性研究 | 药代动力学生物等效性研究 |
| 外推适应证 | 可获得已证实有效性和安全性相似的适应证,在一定条件下,可外推至参照药其他适应证 | 可获得参照药批准的所有适应证 |
注:a生物相似性研究为通过全面的头对头比对试验确定生物类似药与参照药在分子结构、生物学功能、有效性、安全性和免疫原性等方面的相似性;b生物等效性研究为对化学仿制药与参照药在体内以相同速度释放等量活性成分进行的研究
对于新型生物制品来说,研发的关键是获得人体试验的安全性和有效性数据,而生物类似药则是要获得与参照药相似性的全面、完整的证据,具体包括理化特性、生物学活性、纯度和杂质、免疫学特性、质量指标、稳定性,甚至对宿主细胞、制剂处方、规格和内包装材料方面也要进行比对,不一致之处需要给出充分理由。
生物类似药的研发基于逐步递进的原则,依赖全面的比对研究。了解与参照药的生物相似性需要将候选生物类似药与参照药进行全面的头对头的比对研究,包括质量比对研究、非临床比对研究和临床比对研究(图2)。
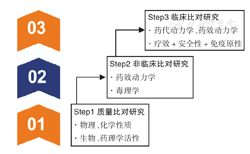
对于生物类似药来说,当其与参照药高度相似、并在一个适应证中进行了全面的安全性和有效性的比对研究时,这些安全性和有效性数据可以外推至参照药的其他适应证,但是外推其他适应证的数据必须有全面比对研究(质量、非临床和临床比对研究)科学证据的支持。外推法的使用基于以下原则,(1)比对研究适应证与外推适应证的作用机制必须相同;(2)比对研究适应证需要选择相对敏感的研究人群:比对研究需在一个足够敏感的适应证人群中进行,以使潜在的临床表现差异能够被检测到;(3)不同临床环境不能使用外推法:当外推适应证的作用机制、药物使用剂量、药代动力学不同时,需单独进行临床试验;(4)外推适应证的安全性和免疫原性数据需要被评估。
对生物类似药的临床疗效要求与参照药相同。在临床比对研究中,通常采用等效性设计,并通过等效性界值的设定来判断研究是否成立。等效性界值一般基于参照药疗效的置信区间进行估算,并结合临床意义进行确定,在不同药物和不同疾病中置信区间不同,此界值是评价生物类似药有效性与参照药相似与否的重要指标。
目前欧洲药品管理局(EMA)已批准上市65个生物类似药,美国食品药品管理局(FDA)已批准上市26个生物类似药。根据随机对照试验(RCT)、系统性综述和Meta分析数据显示,生物类似药的有效性均与参照药相似。
利妥昔单抗是一种人鼠嵌合型的抗CD20单克隆抗体,常用于治疗B细胞恶性肿瘤[如弥漫大B细胞淋巴瘤(DLBCL)、滤泡性淋巴瘤(FL)、慢性淋巴细胞白血病(CLL)等血液肿瘤]和自身免疫性疾病[类风湿性关节炎(RA)、肉芽肿病等][4,5]。
HLX01(商品名:汉利康)是我国首个利妥昔单抗生物类似药,也是我国首个生物类似药,其于2019年2月22日获批上市。在Ⅲ期随机双盲对照头对头临床试验中,共入组407例初治CD20阳性DLBCL患者,分为HLX01联合CHOP(环磷酰胺、多柔比星、长春新碱、泼尼松)方案组和利妥昔单抗参照药联合CHOP方案组,研究的主要终点为治疗6个周期时的客观缓解率(ORR)。治疗6个周期后,HLX01联合CHOP方案组和参照药联合CHOP方案组的ORR分别为94.1%、92.8%,组间ORR差异为1.4%(95%CI 3.59%~6.32%),符合国家药品审评中心(CDE)规定的±12%等效性界值区间,表明HLX01与参照药利妥昔单抗具有临床等效性[6]。
国外一项Meta分析纳入8项利妥昔单抗生物类似药的RCT,共1 534例非霍奇金淋巴瘤(NHL)患者(包括FL、CLL和DLBCL患者),对全球利妥昔单抗生物类似药和参照药进行了临床有效性和安全性的比较[7]。随访4~24周,结果显示,利妥昔单抗生物类似药的ORR与参照药高度相似(RR=1.00,95%CI 0.95~1.05,I2=0),安全性和临床有效性与参照药差异无统计学意义(P>0.05)。
另一项Meta分析从PubMed、Embase、Cochrane Library和Google Scholar数据库检索了头对头RCT,比较了利妥昔单抗生物类似药与参照药的临床疗效[8]。该研究共纳入11项RCT,包含3 163例患者[1 744例RA患者,1 419例NHL患者]。研究的主要终点为临床反应的风险比(RR)。在RA患者中,24周和48周时根据美国风湿病学会评估标准评价的疾病缓解状态相对基线改善20%(ACR20)的集中RR为0.99(P=0.70,95%CI 0.92~1.06)和1.04(P=0.73,95%CI 0.83~1.31);在NHL患者中,24周时ORR的集中RR为1.02(P=0.31,95%CI 0.98~1.07)。表明利妥昔单抗生物类似药在RA和NHL患者中临床疗效类似。
阿达木单抗是一种重组全人源IgG1单克隆抗体,是继英夫利昔单抗(infliximab)和依那西普(etanercept)后,美国FDA批准的第三个肿瘤坏死因子(TNF)抑制剂,主要用于治疗RA、强直性脊柱炎(AS)、斑块状银屑病、幼年特发性关节炎、成年人和儿童克罗恩病、非感染性中间葡萄膜炎、后葡萄膜炎和全葡萄膜炎、银屑病关节炎、溃疡性结肠炎等[9]。
我国目前已有两个阿达木单抗生物类似药获批,分别为广州百奥泰生物制药股份有限公司生产的BAT1406和浙江海正药业股份有限公司生产的HS0106。在与阿达木单抗参照药比对的Ⅲ期随机双盲对照试验中,两个生物类似药研究入组的均为AS急性期患者,研究终点为12周时根据国际脊椎关节炎协会标准评价的疾病缓解状态相对基线改善20%(ASA20),设置的等效性界值区间为±15%。BAT1406的Ⅲ期临床试验入组554例患者,包括BAT1406组(363例)和阿达木单抗参照药组(191例),两组12周时ASA20缓解率分别为75.69%、73.68%,组间缓解率差异为2.16%(95%CI 6.9%~11.22%)[10];HS016的Ⅲ期临床试验入组648例患者,包括HS016组(416例)和阿达木单抗参照药组(232例),12周时ASA20缓解率分别为87.5%、90.1%,组间缓解率差异为-2.59%(90%CI-6.77~1.60)[11],两项研究的组间差异均在有效性界值区间内,表明两款生物类似药的临床疗效与参照药相似,NMPA据此批准二者用于AS、RA和银屑病等自身免疫性疾病的治疗[12,13]。
由上海复星医药生物药平台复宏汉霖公司研发的阿达木单抗生物类似药HLX03的上市申请正在受理中。在2019年欧洲抗风湿病联盟(EULAR)会议上公布了HLX03与阿达木单抗参照药的Ⅰ期临床试验结果。这项随机、双盲、单剂量、平行比对研究证实了HLX03和参照药在药代动力学方面具有生物等效性。HLX03与参照药的最大血药浓度(Cmax)、从给药开始到最后一次可测量浓度的血药浓度-时间曲线下面积(AUClast)的几何均数比值的双侧90%CI在80%~125%内,组间从给药开始到无限时间的血药浓度-时间曲线下面积(AUC0-inf)几何均数比值的双侧90%CI也在等效范围内,进一步证实了两种药物的药代动力学的生物等效性。另外,该项研究证实两药在安全性和免疫原性方面也具有相似性。
在一项对比TNF-α抑制剂生物类似药与参照药的系统性Meta分析中,纳入了9项RCT(包括5个英夫利昔单抗生物类似药、2个阿达木单抗生物类似药和2个依那西普生物类似药),包含3 291例RA和AS患者,以患者临床缓解的RR作为主要终点[14]。RA患者的缓解指标为ACR20和ACR70,AS患者的缓解指标为ASA20。在阿达木单抗生物类似药的两项研究中,与参照药相比,RA患者在12~16周和24~30周ACR20的RR分别为0.98(P=0.86,95%CI 0.82~1.18)和1.01(P=0.91,95%CI 0.90~1.12),表明这两个阿达木单抗生物类似药的临床疗效与参照药类似。同样,英夫利昔单抗生物类似药在RA和AS患者中的临床缓解情况与参照药相似,依那西普生物类似药的临床疗效和不良事件发生率与参照药差异无统计学意义。
另一项研究评估了阿达木单抗生物类似药联合甲氨蝶呤(MTX)对比参照药或安慰剂联合MTX治疗对MTX无响应的活动性RA患者的疗效[15]。该研究采用贝叶斯网络Meta分析法,纳入8项RCT,包含2 543例患者。结果显示阿达木单抗生物类似药组(OR=2.91,95%CI 1.57~5.74)和参照药组(OR=2.80,95%CI 1.81~4.46)的ACR20缓解率明显高于安慰剂组,但是生物类似药组和参照药组的ACR20缓解率差异无统计学意义。基于对严重不良事件的观察,三组在随访12~24周时的安全性无差异,但是仍需长期随访验证。
贝伐珠单抗是一种靶向血管表皮生长因子(VEGF)的重组全人源单克隆抗体,可以抑制内皮细胞生长和血管生成,用于治疗转移性结直肠癌、非鳞状非小细胞肺癌、转移性肾癌、胶质母细胞瘤、子宫颈癌、转移性乳腺癌等[16]。
2019年12月9日NMPA批准上市了由山东齐鲁制药有限公司研发生产的我国首款贝伐珠单抗生物类似药QL1101。在其与参照药的Ⅲ期随机双盲头对头比对试验中,入组了532例局部转移或复发非鳞状非小细胞肺癌患者,临床研究终点为18周时最佳ORR,QL1101组(266例)和贝伐珠单抗参照药组(266例)ORR分别为52.25%、56.02%,组间ORR差异为0.933(90%CI 0.818~1.064),在等效性界值0.75~1.33内,表明QL1101的临床疗效与参照药相似[17]。
在一项旨在比较生物类似药和参照药在肿瘤患者中有效性和安全性的系统性综述及Meta分析中,针对3个生物类似药(利妥昔单抗、贝伐珠单抗和曲妥珠单抗生物类似药),纳入23项RCT[7],其中涉及贝伐珠单抗生物类似药的RCT有6项,包含1 897例患者,使用固定效应模型的合并结果显示,贝伐珠单抗生物类似药的ORR与参照药类似(RR=0.92,95%CI 0.82~1.05,I2=0)。关于贝伐珠单抗生物类似药在肿瘤患者中的无进展生存(PFS)数据有限,只有一项RCT提供了非小细胞肺癌患者的PFS结果,40周时的PFS与参照药相似[加权均数差(WMD)=-0.30,95%CI-3.46~2.86]。
目前大量的研究资料表明,在有效性方面,获批上市的生物类似药与参照药疗效是等同的,这也是生物类似药能被各国药品监管部门批准上市的原因之一。
生物类似药是生物药的一种,虽然相对小分子化学药物,它们的依从性更好,但是有些临床医师担心其免疫原性问题,因为在某些罕见病例中可能导致严重不良反应(如变态反应或迟发性超敏反应),甚至危及患者生命[18]。免疫反应产生的原因往往不只是生产工艺的问题,而是取决于如下的许多因素,(1)产品特性:不恰当的存储或运输过程导致蛋白结构改变或形成多聚体,这可能导致免疫反应;(2)治疗相关因素:给药方式的不同(皮下或静脉给药)、给药时间的不同(连续或间断给药)、合并治疗的影响;(3)患者或疾病相关因素:年龄、基因、免疫状态。
一项系统性研究分析了TNF-α抑制剂和抗CD20单克隆抗体生物类似药的免疫原性[19],该研究共涵盖18个生物类似药,包括阿达木单抗、依那西普、英夫利昔单抗和利妥昔单抗。纳入分析的研究为随机对照头对头临床试验,在依那西普参照药和生物类似药的试验中,抗药抗体(ADA)阳性率(0~13%)和中和抗体(nAb)阳性率(0~3%)最低,阿达木单抗、英夫利昔单抗参照药和生物类似药的ADA阳性率(≤64%)和nAb阳性率(≤100%)最高。但是在依那西普参照药与生物类似药SB4为期52周的比对临床试验中,SB4从基线开始到52周的累积ADA阳性率低于参照药[1%(3/299)比13%(39/296),P<0.001],并且大多数都是短暂的,但是ADA阳性率对参照药组的安全性和有效性未产生影响[20]。该研究显示不同药物ADA阳性率差异大,可能是由于不同研究设置的ADA、nAb检测方法不同导致的,但总体上生物类似药与其参照药的免疫原性无明显差异。
另一项Meta分析显示,依那西普生物类似药的临床疗效和不良事件发生率与参照药无明显差异,而24~30周时ADA阳性率低于参照药(RR=0.05,P<0.000 1,95%CI 0.01~0.21)[14]。大多数研究在补充材料中报道了ADA对药物代谢、药代动力学、安全性和有效性的影响。通常,ADA阳性患者比阴性患者体内的药物浓度更低,药物清除率更高,但是对有效性无影响,与参照药是相当的。
由于我国生物类似药上市时间较短,暂无相关上市后安全性数据,但是过去十年,在欧盟的安全监控系统中尚未发现生物类似药与参照药之间存在不良反应性质、严重程度或发生频率的差异。一篇文章综述了已发表的上市前和上市后药物的相关研究,总结了治疗RA生物类似药的安全性;其评估的生物类似药包括TNF抑制剂(阿达木单抗、依那西普和英夫利昔单抗)和抗CD20单克隆抗体[21]。从检索到的研究数据来看,生物类似药的安全性与其参照药类似[22,23,24,25,26](表2)。对于在2013年全球第一个获批的单克隆抗体生物类似药CT-P13来说,前瞻性随机临床试验和上市后的研究均未发现新的安全性事件。目前为止,TNF抑制剂或利妥昔单抗生物类似药在治疗RA时,也未出现新的安全性事件。
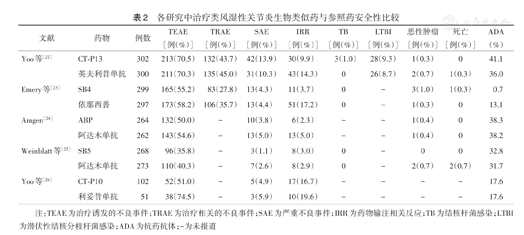
各研究中治疗类风湿性关节炎生物类似药与参照药安全性比较
各研究中治疗类风湿性关节炎生物类似药与参照药安全性比较
| 文献 | 药物 | 例数 | TEAE[例(%)] | TRAE[例(%)] | SAE[例(%)] | IRR[例(%)] | TB[例(%)] | LTBI[例(%)] | 恶性肿瘤[例(%)] | 死亡[例(%)] | ADA(%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Yoo等[22] | CT-P13 | 302 | 213(70.5) | 132(43.7) | 42(13.9) | 30(9.9) | 3(1.0) | 28(9.3) | 1(0.3) | 0 | 41.1 |
| 英夫利昔单抗 | 300 | 211(70.3) | 135(45.0) | 31(10.3) | 43(14.3) | 0 | 26(8.7) | 2(0.7) | 1(0.3) | 36.0 | |
| Emery等[23] | SB4 | 299 | 165(55.2) | 83(27.8) | 13(4.3) | 11(3.7) | 0 | - | 3(1.0) | 1(0.3) | 0.7 |
| 依那西普 | 297 | 173(58.2) | 106(35.7) | 13(4.4) | 51(17.2) | 0 | - | 1(0.3) | 0 | 13.1 | |
| Amgen[24] | ABP | 264 | 132(50.0) | - | 10(3.8) | 6(2.3) | - | - | 1(0.4) | 0 | 38.3 |
| 阿达木单抗 | 262 | 143(54.6) | - | 13(5.0) | 13(5.0) | - | - | 1(0.4) | 0 | 38.2 | |
| Weinblatt等[25] | SB5 | 268 | 96(35.8) | - | 3(1.1) | 8(3.0) | 0 | - | 0 | 0 | 32.8 |
| 阿达木单抗 | 273 | 110(40.3) | - | 7(2.6) | 8(2.9) | 0 | - | 2(0.7) | 2(0.7) | 31.7 | |
| Yoo等[26] | CT-P10 | 102 | 52(51.0) | - | 5(4.9) | 17(16.7) | - | - | - | - | 17.6 |
| 利妥昔单抗 | 51 | 38(74.5) | - | 3(5.9) | 10(19.6) | - | - | - | - | 17.6 |
注:TEAE为治疗诱发的不良事件;TRAE为治疗相关的不良事件;SAE为严重不良事件;IRR为药物输注相关反应;TB为结核杆菌感染;LTBI为潜伏性结核分枝杆菌感染;ADA为抗药抗体;-为未报道
对于生物类似药在外推适应证中使用的安全性问题也是临床医师担心的方面。由于外推法是基于科学证据才能施行的,所以具有可靠性。CT-P13是英夫利昔单抗生物类似药,根据PLANETRA和PLANETAS试验结果,CT-P13获批用于其参照药的所有适应证,包括克罗恩病、炎症性肠病(IBD)、斑块性银屑病和银屑病性关节炎,其中IBD是在考察了CT-P13物理化学、生物学数据和非比对性观察研究后获批的。韩国的一项药物上市后的研究入组了173例接受CT-P13治疗的IBD患者,30周时的不良事件发生率仅为22.0%,治疗相关不良事件发生率仅为10.4%,无恶性肿瘤发生[27]。意大利一项入组397例IBD患者的观察性研究显示,CT-P13治疗后严重不良事件发生率为8.3%,输注相关反应发生率为5.3%[28]。
因此,生物类似药的安全性(包括不良事件发生率、免疫原性及外推适应证等)与参照药都是相似的,可以被安全地用于临床实践。
药物转换是指由处方医生决定将一种药物转换为另一种治疗用途相同的药物。对于生物类似药来说,由于和参照药没有临床结果的差异,而治疗费用更低、可及性更好,所以也是临床药物转换的选择之一。目前临床医师对生物类似药的转换主要有两种情况,一种是生物类似药和参照药之间的转换,另一种是生物类似药之间的转换。
已有研究表明,从参照药转换到对应的生物类似药不会产生任何新的安全性事件。EGALITY是一项分析依那西普参照药和生物类似药之间多轮转换安全性和有效性的研究,入组了慢性斑块型银屑病患者,其中200例患者接受3轮生物类似药与参照药之间的转换治疗,其他患者保持参照药治疗;结果显示两组治疗相关不良事件相似,转换组的ADA发生率较低且短暂,没有检测到nAb[29]。另一项系统性综述研究纳入英夫利昔单抗参照药转换为生物类似药CT-P13治疗IBD的29项研究,结果显示在诱导和维持治疗阶段,CT-P13的安全性和有效性均与参照药相似,从参照药转换为CT-P13的有效性和安全性不劣于一直使用CT-P13[30]。
在真实世界中,也有患者转换为生物类似药后又换回参照药的情况。一项来自丹麦的观察性队列研究是基于丹麦DANBIO数据库注册研究后的真实世界研究,其对比依那西普参照药转换为生物类似药SB4与参照药继续治疗的效果,研究入组2 061例RA、银屑病关节炎、轴性脊椎关节炎患者,其中1 621例(79%)患者接受了转换为SB4的治疗。疾病活动状态的改变率在转换前3个月和转换后3个月无差异[RA患者28个关节疾病活动评分(ΔDAS28)≥0.6,22%比24%]。1年内一直使用SB4患者保留率为77%,一直使用参照药患者保留率为83%。在随访过程中,有120例(7%)SB4治疗患者转换为参照药,但是转换后临床特征与未转换回参照药类似,发生转换的多数原因是患者主观选择和非特异性的药物反应[31]。
目前NMPA对临床药物转换暂无具体规定,主要由医生的临床经验决定。欧盟由各成员国自行决定,但芬兰、荷兰、德国和挪威的国家监管机构已经声明,批准上市的生物类似药在临床上可以和参照药互换使用[32]。一些大型综述性分析研究显示,停止生物类似药的使用概率在不同研究中差别较大,从0~85%不等,表明生物类似药转换在不同患者中的异质性较大,因此临床上生物类似药转换的决定应由临床医师根据患者情况并基于全面的科学证据而做出[33]。
因此,基于NMPA对临床药物转换暂无具体规定的情况,参考其他国家的规定,建议主要由临床医生根据临床实际及患者经济情况决定,目前资料显示转换用药是安全的。
生物药的出现极大地改善了不能治愈疾病的预后,但是由于价格昂贵,其在临床的使用受到很大限制。随着许多原研生物药的专利到期,高性价比的生物类似药已陆续被研发并获批上市,使得更多患者能够及时接受治疗。
一般而言,生物类似药以低于其参照药的价格推向市场,在增加市场竞争的同时,可以降低复杂医疗条件下的总体成本。英国一项分析生物类似药引入市场后对国家健康系统支出影响的研究中,根据研究应用的预测模型显示,在未来三年内,生物制品的开支将减少4 400万英镑。随着生物类似药的引入,预计到2020年累计可节省2.85亿英镑[34]。由此可见,生物类似药的发展有助于患者和社会真正获益,但是鉴于我国生物类似药研发时间较短、上市药物较少的状况,其发展仍需一定时间。
执笔人 马军、朱军、黄慧强、赵维莅
组长 马军、朱军、沈志祥、石远凯、黄晓军、吴德沛
副组长 黄慧强、邱录贵、李建勇、赵维莅、李小秋、张清媛
秘书长 宋玉琴、郭晔、李志铭、赵东陆、张岩
专家组成员(按姓氏汉语拼音排序)白鸥(吉林大学白求恩第一医院)、鲍慧铮(吉林省肿瘤医院)、曹军宁(复旦大学附属肿瘤医院)、丁凯阳(安徽省肿瘤医院)、范艳玲(哈尔滨血液病肿瘤研究所)、冯继锋(江苏省肿瘤医院)、高子芬(北京大学医学部)、高怡瑾(上海儿童医学中心)、贡铁军(哈尔滨血液病肿瘤研究所)、郭晔(同济大学附属东方医院)、郝文鹏(哈尔滨血液病肿瘤研究所)、胡建达(福建医科大学附属协和医院)、黄慧强(中山大学肿瘤防治中心)、黄晓军(北京大学人民医院)、靳凤艳(吉林大学白求恩第一医院)、金洁(浙江大学医学院附属第一医院)、李建勇(南京医科大学第一附属医院 江苏省人民医院)、李晓玲(辽宁省肿瘤医院)、李小秋(复旦大学附属肿瘤医院)、李艳(中国医科大学附属第一医院)、李志铭(中山大学肿瘤防治中心)、梁红(哈尔滨血液病肿瘤研究所)、刘丽宏(河北医科大学第四医院)、刘澎(复旦大学附属中山医院)、刘卫平(北京大学肿瘤医院)、刘卓刚(中国医科大学附属盛京医院)、马军(哈尔滨血液病肿瘤研究所)、牛挺(四川大学华西医院)、邱录贵(中国医学科学院血液病医院 中国医学科学院血液学研究所)、苏丽萍(山西省肿瘤医院)、双跃荣(江西省肿瘤医院)、宋永平(河南省肿瘤医院)、宋玉琴(北京大学肿瘤医院)、石远凯(中国医学科学院肿瘤医院)、沈建箴(福建医科大学附属协和医院)、沈志祥(上海交通大学医学院附属瑞金医院)、王巍(哈尔滨医科大学附属第二医院)、王欣(山东省立医院)、王志国(哈尔滨血液病肿瘤研究所)、吴德沛(苏州大学附属第一医院)、徐卫(南京医科大学第一附属医院 江苏省人民医院)、杨顺娥(新疆医科大学附属肿瘤医院)、赵东陆(哈尔滨血液病肿瘤研究所)、赵维莅(上海交通大学医学院附属瑞金医院)、张明智(郑州大学第一附属医院)、张会来(天津医科大学肿瘤医院)、张清媛(哈尔滨医科大学附属肿瘤医院)、张曦(陆军军医大学新桥医院)、张翼鷟(中山大学肿瘤防治中心)、朱军(北京大学肿瘤医院)、周道斌(北京协和医院)、周剑峰(华中科技大学同济医学院附属同济医院)、周辉(湖南省肿瘤医院)
所有作者均声明不存在利益冲突
























