
系统评价C-KIT基因突变与儿童核心结合因子相关急性髓系白血病(CBF-AML)预后的关系。
以"KIT""Acute Myeloid Leukemia""Children"为英文检索词检索PubMed数据库;以"KIT""急性髓系白血病""儿童"为检索词检索中国期刊全文数据库(CNKI)、中国生物医学文献数据库(CBM)、维普数据库、万方数据库,检索时间为建库至2020年10月1日。文献经过严格筛选后纳入分析;根据基因检测明确有无基因改变,分为C-KIT突变组和野生组,分析两组完全缓解(CR)率、无事件生存(EFS)率、总生存(OS)率的差异。
共纳入6篇文献,其中英文文献4篇,中文文献2篇,共667例患者。儿童CBF-AML患者中,C-KIT突变组与野生组EFS率比较,差异有统计学意义(HR=2.40,95% CI 1.47~3.89,P=0.001);两组CR率和OS率差异均无统计学意义(OR=0.93,95% CI 0.48~1.80,P=0.830;HR=1.92,95% CI 0.96~3.83,P=0.065)。
C-KIT基因突变可能是儿童CBF-AML患者预后不良的危险因素。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
核心结合因子相关急性髓系白血病(CBF-AML)是存在异常染色体t(8;21)(q22;q22)和inv(16)/ t(16;16)(p13;q22)的一种白血病亚型,其相应形成的融合基因为RUNX1-RUNX1T1和CBFβ-MYH11。CBF-AML是儿童AML中最常见的一种亚型,发生率20%~22%[1,2,3]。与其他核型异常相比,CBF-AML在强化诱导化疗后完全缓解(CR)率约90%,在英国医学研究委员会(MRC)、美国国家综合癌症网络(NCCN)、欧洲白血病网络(ELN)及相关研究中,CBF-AML被认为是预后良好的AML亚组[4,5,6]。但仅28%~70% CBF-AML患者长期生存,提示可能存在未知危险因素[7,8,9,10]。C-KIT是由造血干细胞表达的Ⅲ型受体酪氨酸激酶,通过与干细胞因子(SCF)配体结合,C-KIT受体磷酸化,激活细胞内信号转导途径,影响细胞的生长和凋亡[11,12]。作为CBF-AML中最常见的突变基因之一,成人CBF-AML尤其是伴t(8;21)的患者,C-KIT基因突变与不良预后相关[1,5,13,14,15];20%~40%儿童CBF-AML患者出现C-KIT突变[1,3,16],但关于C-KIT突变与儿童CBF-AML预后的相关性尚存在争议[1,3,13,17,18,19]。部分研究显示C-KIT基因突变会有更差的无事件生存(EFS)、总生存(OS)等[17,18,19]。为了对患儿更好地进行危险度分级,实现诊疗的个体化,并根据预后分级给予个体化治疗,有效提高患儿治疗缓解率及生命质量、延长OS时间,本研究对C-KIT突变与儿童CBF-AML预后的关系进行Meta分析,探讨C-KIT突变在儿童CBF-AML预后评估中的价值。
以"KIT""Acute Myeloid Leukemia""Children"为英文检索词检索PubMed数据库;以"KIT""急性髓系白血病""儿童"为检索词,检索中国期刊全文数据库(CNKI)、中国生物医学文献数据库(CBM)、维普数据库、万方数据库;检索时间为建库至2020年10月1日。语种为中文及英文。
纳入标准:(1)2020年10月1日前已发表的关于C-KIT突变与儿童CBF-AML预后关系研究的相关文献;(2)通过骨髓形态学、免疫学、细胞遗传学及分子生物学诊断CBF-AML的儿童患者;(3)直接提供或可间接推算获得C-KIT基因突变型和野生型患者EFS率及OS率的HR值及其95%置信区间(CI),数据中至少1项预后指标;(4)原始全文可查阅;(5)有完整的研究结果。排除标准:(1)复发或继发患者;(2)成年人CBF-AML;(3)文献研究数据不完整,与作者联系后无法获得所需数据;(4)原始文献研究对象为非人类;(5)重复发表文献;(6)综述文献;(7)各种原因无法获得全文及不能满足Cochrane手册要求的文献。
(1)浏览数据库文献的标题和摘要,初步选取相关文献;(2)阅读全文,进一步选取与研究相关的文献;(3)根据文献纳入与排除标准,严格筛选,确定最终纳入文献;(4)选取纽卡斯尔-渥太华量表[20](NOS)为评价标准。具体评分项目包括研究对象选择、可比性、暴露评价或结局评价。NOS对文献质量的评价采用了半量化的评分规则,满分为9分,质量可分为3个等级,5分以上可纳入分析;(5)对纳入的文献进行数据提取,内容包括第一作者、发表年份、患者人数及所在国家或地区、年龄、CR率、OS率、EFS率;(6)文献纳入与排除过程由两位研究者独立完成,在达成共识前,不进行进一步分析。如有必要,请第3位调查员发表意见。
对符合纳入标准的文献数据归纳整理后采用RevMan5.2、STATA12.0软件进行定量分析。对纳入的研究进行异质性检验,I2值越大则异质性越大,I2≥50%使用随机效应模型,I2<50%使用固定效应模型。同时对合并的研究结果进行敏感性分析,并通过Begg秩相关法及Egger线性回归评价所纳入研究的发表偏倚。检验水准α=0.05。
共检索394篇文献,排除重复及不相关的文献后,最终纳入6篇文献。研究对象均为儿童CBF-AML患者,共667例,其中突变168例,纳入文献的基本特征见表1。
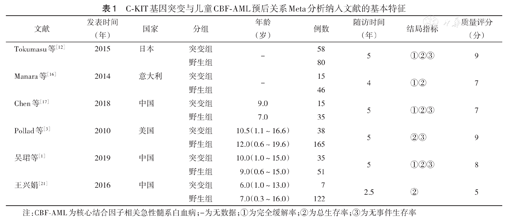
C-KIT基因突变与儿童CBF-AML预后关系Meta分析纳入文献的基本特征
C-KIT基因突变与儿童CBF-AML预后关系Meta分析纳入文献的基本特征
| 文献 | 发表时间(年) | 国家 | 分组 | 年龄(岁) | 例数 | 随访时间(年) | 结局指标 | 质量评分(分) |
|---|---|---|---|---|---|---|---|---|
| Tokumasu等[12] | 2015 | 日本 | 突变组 | - | 58 | 5 | ①②③ | 9 |
| 野生组 | 80 | |||||||
| Manara等[16] | 2014 | 意大利 | 突变组 | - | 15 | 4 | ①② | 7 |
| 野生组 | 46 | |||||||
| Chen等[17] | 2018 | 中国 | 突变组 | 9.0 | 15 | 5 | ①②③ | 7 |
| 野生组 | 7.0 | 35 | ||||||
| Pollad等[3] | 2010 | 美国 | 突变组 | 10.5(1.1~16.6) | 38 | 5 | ②③ | 9 |
| 野生组 | 12.0(0.6~19.6) | 165 | ||||||
| 吴珺等[1] | 2019 | 中国 | 突变组 | 10.0(1.0~15.0) | 35 | 5 | ①②③ | 8 |
| 野生组 | 9.0(0.6~15.0) | 51 | ||||||
| 王兴娟[21] | 2016 | 中国 | 突变组 | 6.0(1.0~13.0) | 7 | 2.5 | ② | 5 |
| 野生组 | 7.0(0.3~16.0) | 122 |
注:CBF-AML为核心结合因子相关急性髓系白血病;-为无数据;①为完全缓解率;②为总生存率;③为无事件生存率
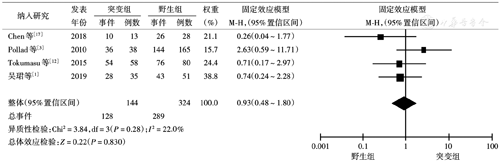
4篇研究评估C-KIT突变对EFS的影响,无明显异质性(I2=22.0)(图1),突变组及野生组在CR中差异无统计学意义(P=0.830)。同时使用Begg's和Egger's检验评估发表偏倚(Begg's P=1.000,Egger's P=0.744),故不存在发表偏倚。
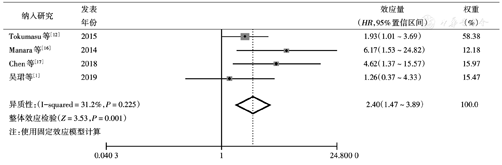
4篇研究评估C-KIT突变对EFS的影响,无明显异质性(I2=31.2),结果提示野生组EFS高于突变组,差异有统计学意义(P=0.001)(图2);无发表偏倚(Begg's P=0.734,Egger's P=0.465)。
6个研究评估C-KIT突变对OS的影响,合并后结果显示野生组OS优于突变组,差异有统计学意义(P=0.020);同时研究之间存在异质性(P=0.065,I2=51.8%)(图3),通过敏感性分析(图4),寻找异质性主要来源;在王兴娟[21]研究内容中,突变组随访时间约为30个月,其他研究均在5年左右。将该研究剔除后,异质性降低(P=0.498,I2=0),合并后结果显示,突变组及野生组间OS差异无统计学意义(P=0.065)。
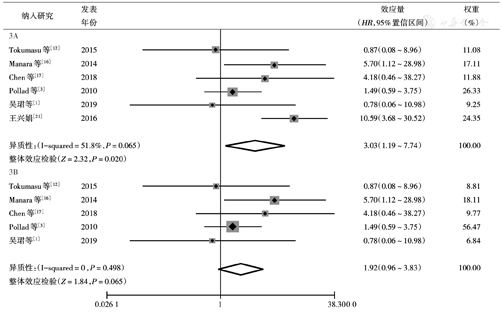
注:3A采用随机效应模型计算;3B采用固定效应模型计算
AML是儿童常见的血液系统恶性疾病之一,以未成熟的前体细胞分化停滞、恶性克隆增殖、浸润其他器官或组织,并抑制正常骨髓造血为特点。其主要包括Ⅰ类和Ⅱ类基因突变。Ⅰ类基因突变指激酶信号通路基因的异常激活,例如NRAS、FLT3、C-KIT和PTPN1,这类突变导致细胞永生和增殖,表现出在低配体浓度下完全激活的酪氨酸激酶活性。Ⅱ类基因突变指转录因子失活,如PML/RARA、CBFB/MYH11、RUNX1/RUNX1T1和CEBPA突变等,表现出不依赖配体的酪氨酸激酶活性,最终造成造血干细胞异常分化,产生白血病细胞[22,23]。
CBF-AML是AML中最常见的一种亚型,发生率20%~22%,其主要特征为存在t(8;21)(q22;q22)和inv(16)/t(16;16)(p13;q22)异常染色体,形成融合基因RUNX1-RUNX1T1和CBFβ-MYH11。CBF是正常造血中必需的异二聚体转录因子,其每个α-亚基和β-亚基由RUNX家族和CBFB基因编码,在CBF-AML的异常核型中产生嵌合蛋白,破坏CBF复合物并抑制转录激活[24,25,26],造成造血干细胞异常分化,进而产生白血病细胞。RUNX1-ETO蛋白通过与C-KIT启动子结合并将组蛋白乙酰转移酶P300募集到C-KIT启动子而表观遗传地反式激活C-KIT,导致其突变,从而导致C-KIT基因活化增加[25]。因此在CBF-AML中,C-KIT突变较其他核型高[1,2,3]。
C-KIT是由造血干细胞表达的Ⅲ型受体酪氨酸激酶,位于染色体4q11-12上,在急性髓系白血病、骨髓增生异常综合征和肥大细胞增多症均有突变相关报道。该基因突变导致了活化级联的细胞因子依赖性的丧失,导致自主、独立细胞因子激酶激活和无限制的细胞增殖[27,28,29,30]。
本研究对携带C-KIT突变的儿童CBF-AML患者进行分析,结果显示C-KIT基因突变对于患儿CR及OS差异均无统计学意义(均P>0.05);对于长期预后指标EFS差异有统计学意义(P<0.05),表明C-KIT基因突变对于患儿远期的生存可能存在不利影响。产生这种不良预后的结果,可能与C-KIT基因的功能相关。C-KIT是一种调节髓系造血的重要基因,其编码的受体蛋白即CD117,可被干细胞因子(SCF)激活。CD117蛋白主要分布在造血干细胞及祖细胞的表面,通过与其配体SCF特异性结合,构成SCF-C-KIT信号通路,对血液祖细胞增殖及凋亡的调控作用[11,12,31]。当C-KIT基因的异常表达时,CD117即使在未与SCF结合的情况下仍能自发性地磷酸化,使SCF-C-KIT信号通路出现不受控制的持续性激活状态[32,33],而SCF-C-KIT信号通路出现异常活化时,会导致造血细胞恶性增殖和分化紊乱,从而导致血液肿瘤的发生和发展。目前在C-KIT突变的研究中,多集中在外显子8(编码C-KIT的胞外域)和17(编码C-KIT的激活环)上,少有研究该基因的其他区域,包括外显子10和11(分别编码跨膜结构域和近膜结构域)[11,12,17];当突变位于外显子8和17时,其预后较突变位于外显子11的患儿差[11],因此C-KIT基因突变所致的预后差异还需进一步分析不同的突变区域,评估不同突变区域的风险性。
目前对于酪氨酸激酶受体基因突变的治疗,多以靶向治疗为主,如伊马替尼等[18,34]。C-KIT是一种酪氨酸激酶受体基因,与其他的Ⅲ型受体酪氨酸激酶家族的其他成员具有相同的结构,结合本研究提示该基因突变提示存在不利影响,因此当发现该基因突变时应于早期加入酪氨酸激酶抑制剂进行治疗。
由于本文可纳入的原始研究及研究数据有限,无法进一步进行亚组分析评估各因素影响的差异,如不同年限生存率、不同突变区域、人种差异等;同时原始研究中也可能存在失访偏倚。这些因素均可能使分析结果的准确性受影响,故本研究存在一定的局限性,有待进一步开展大规模、高质量的研究试验。今后的研究应注意扩大样本量,考虑不同的突变区域对预后的影响,为临床预后判断、危险度分层及用药指导提供依据。
所有作者均声明不存在利益冲突

























