
本综述阐述了神经母细胞瘤候选致病基因的研究进展,并对这些基因的功能进行归类总结。同时也回顾了目前用于寻找该病致病或易感基因的各种方法,分析了其优缺点,并创新性地提出靶向捕获的二代测序技术是寻找神经母细胞瘤潜在致病突变基因的关键方法,或许可以为治疗干预措施提供潜在的新靶点。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
神经母细胞瘤(neuroblastoma,NB)是起源于原始神经嵴细胞的胚胎性肿瘤,为儿童最常见的颅外实体瘤。NB具有高度异质性,能从自行消退到病情迅速进展,这必然与其独特的分子生物学复杂性密切相关[1,2]。近些年来,已经有越来越多的报道关注与NB发病进展相关的基因改变,但NB的发病机制尚未明确。靶向捕获的二代测序技术具有快速、高通量、高特异性以及低成本的特点[3],因此可被用于探究NB的致病或易感基因。
目前,此领域多数相关研究基于全基因组关联分析、基因芯片技术以及二代基因测序技术。根据有无遗传史,NB可分为遗传性和散发性两种类型,两者均是遗传因素和环境因素相互作用的结果。
遗传性NB主要表现为某一基因的罕见变异,仅占所有NB患儿的1%~2%,为不完全外显的常染色体显性遗传[4]。目前已经报道的关于遗传性NB的种系突变主要发生在两个基因中,即配对样同源异形盒2B基因(PHOX2B)和间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)基因。PHOX2B基因是第一个被证实和NB相关的基因,所有NB中约有1%是由PHOX2B的突变导致的[4,5]。该基因位于人类染色体的4p12区域,编码调控神经嵴发育的转录因子。该基因以功能丢失突变的方式影响NB细胞终末分化而致病[6]。PHOX2B突变绝大多数都发生在伴有合并症的个体中(包括先天性中枢性低通气综合征、先天性巨结肠、多发性神经纤维瘤和嗜铬细胞瘤等),而这些伴有合并症的患儿约占遗传性NB的10%[5]。ALK基因突变在遗传性NB中较PHOX2B基因突变更常见。ALK基因位于人类染色体2p23,编码酪氨酸激酶受体,该蛋白属于胰岛素受体超家族。它对大脑以及神经系统中特定神经元的发育发挥着重要作用。ALK异常作用机制为ALK扩增或突变,使ALK磷酸化增加而致激酶活性增高,最终导致肿瘤的发生[7]。在遗传性NB中,ALK基因是由于编码氨基酸序列的区域(如F1174、F1245和R1275等热点区域)发生突变而致病[8,9,10,11,12,13,14]。
散发性NB也是一种复杂的基因疾病,不同于遗传性NB的是,散发性NB是由于多形态的等位基因共同改变导致NB的发生。19世纪80年代高水平MYCN基因扩增的发现,揭开了将肿瘤特异性基因组改变作为NB预后指标的序幕[15,16]。自此,除了MYCN基因扩增以外,其他与散发性NB发生密切相关的基因改变也逐渐被人们所认识,例如LMO1、LIN28B等易感基因,染色体17q获得以及1p、3p或者11q的缺失等,ALK突变或扩增、ATRX突变等。
回顾大量NB全基因组关联分析(GWAS)相关文献,总结出与NB发生密切相关的易感基因分为两类,第一类基因改变发生于高危组NB患者中,包括CASC15、BARD1、LMO1、LIN28B和HACE1基因[17];第二类基因改变发生于低危组NB患者中,包括DUSP12、DDX、IL31RA以及HSD17B12基因[18]。其中,LMO1基因是近些年来研究最热的NB易感基因[19]。Wang等[20]发现该基因与NB相关的5个重要的SNP为rs4758051、rs10840002、rs110419、rs204938和rs110420。而北京儿童医院明确rs204926与NB的关系最密切[17]。LMO1基因编码的蛋白为转录因子,通过竞争性结合到具有特定DNA结合区的转录因子上来间接调控转录。以前也有研究表明LMO1参与神经系统发育的监管,以及LMO1启动子能增强中枢神经系统的基因表达[20]。
众多研究表明,染色体结构和数目改变对NB的预后影响有着明显的差异。Janoueix-Lerosey等[21]用比较基因组杂交(CGH)技术分析了近500例NB患者,发现完整染色体拷贝数变异的肿瘤有着极好的生存率,相反,具有任何类型的节段性染色体改变的肿瘤患者复发风险较高。最终,其认为在发生节段性染色体改变的NB患者中,4期、年龄、MYCN扩增、1p和11q缺失以及1q获得是不良预后的独立预测因子。
(1)1p缺失1号染色体短臂的部分缺失在30%NB中可观察到,某些缺失与MYCN扩增有关。据报道,伴有MYCN基因扩增的缺失片段往往比无MYCN基因扩增的缺失片段大(伴MYCN扩增的1p中位缺失片段大小为84Mb,不伴MYCN扩增的中位缺失片段大小为46Mb)[22]。目前发现1p缺失与NB发病或预后相关的基因为位于1p36.31的CAMTA1和位于1p36.2的PIK3CD[23,24]。
(2)11q缺失11号染色体整个缺失占散发性NB的15%左右,部分缺失占22%左右。11q的缺失在MYCN未扩增的NB中更常见[22]。Carén等[25]从165例NB患者的SNP芯片技术检测以及随访调查中发现,伴有11q缺失提示预后不良,甚至等同于MYCN基因扩增,11q缺失涉及到的基因包括CADM1和ATM。CADM1为抑癌基因,其失活导致肿瘤细胞入侵或者转移。ATM基因编码PI3/PI4蛋白家族,为细胞周期检测点激酶,在修复DNA损伤或者稳定基因组方面发挥重要作用。
(3)17q获得目前大量研究已经明确17q获得对NB的影响,而且这种改变在NB中比较常见。其中涉及到的基因有BIRC5、NME1和PPM1D等[26]。Jeison等[27]发现染色体11的异位是导致17q获得的原因[der(11)t(11q;17q)]。BIRC5基因为凋亡抑制(IAP)基因家族成员之一,其编码负性调控蛋白,抑制凋亡细胞的死亡。NME1基因为转移抑制基因,可下调参与转移过程的靶向基因。PPM1D基因编码PP2C蛋白家族,是细胞应激反应途径的负调控因子。
(4)9p和3p缺失Carén等[22]用SNP芯片分析NB样本时发现染色体9p会发生纯合或杂合性缺失,涉及到的基因包括CDKN2A和CDKN2B。还可见到3p的纯合性缺失,涉及到的基因包括RBMS3和LSAMP基因。CDKN2A和CDKN2B基因均位于9p21,是CDK激酶抑制剂,参与细胞周期调控,为抑癌基因。RBMS3基因编码RNA结合蛋白,属于单链c-myc基因结合蛋白家族,这些蛋白的作用是通过与C-MYC蛋白相互作用来调控DNA复制、转录、凋亡以及细胞周期[28]。
与成人恶性肿瘤相比,儿童恶性肿瘤体细胞突变频率较低[29]。NB的外显子突变频率仅为每个Mb单位0.2~0.4。突变频率与肿瘤分期密切相关,较低期的NB患儿体细胞突变发生的可能性较小[30]。除了MYCN基因,ALK基因在体系突变中仍然占主要地位[13,30,31,32]。其次为ATRX基因,占NB体系突变的2.5%左右,但通常发生在高龄NB患儿。其他基因体系突变频率均较低[13,30,32]。
(1) MYCN基因位于染色体2p24的MYCN基因的扩增已被明确视为NB预后不良因素之一,被纳入国际预后评价指标[15]。作为NB发生和引起肿瘤细胞治疗抵抗的重要致癌基因,其最主要的作用是缩短细胞周期,促进细胞增殖,抑制细胞分化、凋亡。MYCN扩增(10倍的二倍体基因组的复制或>4倍的与信号相关的2号染色体的复制)在NB的总体发生率大约有22%,而且大多与其他危险因素同时存在[27]。例如,MYCN基因往往也可伴随其他基因共同扩增,包括MEIS1,DDX1、NBAS和ODC1[33,34]。进一步研究发现,约1%的NB存在除了MYCN扩增的其他位点的扩增,这些位点集中在染色体19p12,2p25,2p23,21q21,22q11和12q13-14,涉及到的基因有CDK4、CDK6、MDM2、CCND1等[35]。这些基因的扩增提示我们除了MYCN基因以外,还应关注与NB发生相关的其他扩增基因。最近Pugh等[13]还发现在无MYCN基因扩增的NB患者中还可发生MYCN突变(p.Phe44Leu)。
(2)ALK基因除了在遗传性NB中ALK基因与NB的关系已得到明确,研究人员发现ALK基因活化体系突变或扩增在散发性NB中也起重要作用。约8%~10%的散发性NB存在ALK的活化体系突变,突变簇集中在外显子23和25(F1174和R1275),即编码酪氨酸激酶受体的区域[26,33],而且大量Meta分析还报道F1174L突变与MYCN基因扩增有关[33,36,37];ALK基因扩增可伴随MYCN、DDX1和NAG基因的扩增[26]。还有报道发现并不是所有ALK基因扩增均会导致NB的发生,距离ALK仅有13Mb的MYCN基因如果与ALK同时扩增,则更容易导致散发性NB的发生[11,38]。
(3)ATRX基因ATRX基因位于染色体Xq13.1-q21.1,参与染色质重排、细胞分化和DNA修复。据Kurihara等[39]报道ATRX发生的体细胞突变为外显子5~10丢失、错义突变(Q929E和A1690D)及无义突变(E555*)。同时还发现位于染色体6p21.3的另一基因DAXX也与ATRX功能类似,可发生框移突变A470 indel,二者突变均致端粒加长,而端粒加长往往也是肿瘤发生的一个重要机制。ATRX的突变与NB发病年龄密切相关,年龄小于18个月的4期NB患儿中未检出ATRX突变,18个月到12岁之间突变的发生率为17%,而大于12岁发病的患者中这一突变率升高到44%,但诊断年龄与ATRX突变之间的关系及意义还需要进行大量的实验研究[30,32]。
(4)ARID1A和ARID1B基因位于染色体1p35.3的ARID1A基因和6q25.1的ARID1B基因,它们所编码的蛋白质产物均为SWI/SNF家族成员,作用是通过改变某些基因周围的染色质结构来调控这些基因的表达,所以这两个基因与细胞分化、染色质重塑相关。Sausen等[31]检测到ARID1A基因可发生体细胞无义、错义、截断突变以及杂合性缺失(LOH),ARID1B基因可发生体细胞杂合性缺失以及点突变,并认为它们的变异可以导致NB患者早期治疗失败以及生存率的降低。另外根据报道,涉及到染色体重塑的其他基因,如组蛋白乙酰化转移酶基因EP300和CREBBP,染色质解旋酶DNA结合基因CHD9[40],组蛋白去甲基化酶基因KDM5A和染色质重塑锌指基因IKZF1也均会发生体细胞突变,参与NB的发生[33]。
NB的发生是一个多级过程,可同时激活多种致癌信号通路。首先,控制神经前体细胞增殖、分化及存活的相关信号通路,如Wnt、Notch及Hedgehog[41]。以Notch信号为例,当抑制信号后,可以使NB细胞向更成熟的方向分化[42]。在神经系统发育的早期阶段,Notch信号通路可以调节细胞的增殖、分化及凋亡,也可通过与PI3K-AKT信号关联,参与肿瘤血管的生成[43];其次,在众多肿瘤形成中均起关键作用的酪氨酸激酶受体介导的RAS-MARK和PI3K-AKT信号通路也调控NB的形成。这些信号通路通过影响细胞的凋亡、增殖、DNA修复以及肿瘤细胞的转移、血管形成等机制致癌[44,45]。Eleveld等[46]还报道了在复发NB中,RAS-MARK信号通路相关的基因突变现象较常发生。(表1)
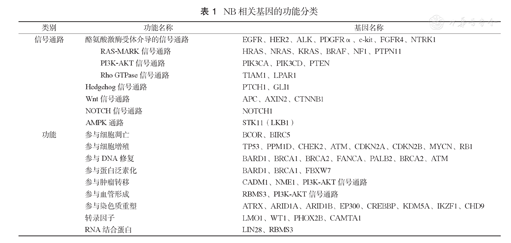
NB相关基因的功能分类
NB相关基因的功能分类
| 类别 | 功能名称 | 基因名称 | |
|---|---|---|---|
| 信号通路 | 酪氨酸激酶受体介导的信号通路 | EGFR、HER2、ALK、PDGFRα、c-kit、FGFR4、NTRK1 | |
| RAS-MARK信号通路 | HRAS、NRAS、KRAS、BRAF、NF1、PTPN11 | ||
| PI3K-AKT信号通路 | PIK3CA、PIK3CD、PTEN | ||
| Rho GTPase信号通路 | TIAM1、LPAR1 | ||
| Hedgehog信号通路 | PTCH1、GLI1 | ||
| Wnt信号通路 | APC、AXIN2、CTNNB1 | ||
| NOTCH信号通路 | NOTCH1 | ||
| AMPK通路 | STK11(LKB1) | ||
| 功能 | 参与细胞凋亡 | BCOR、BIRC5 | |
| 参与细胞增殖 | TP53、PPM1D、CHEK2、ATM、CDKN2A、CDKN2B、MYCN、RB1 | ||
| 参与DNA修复 | BARD1、BRCA1、BRCA2、FANCA、PALB2、BRCA2、ATM | ||
| 参与蛋白泛素化 | BARD1、BRCA1、FBXW7 | ||
| 参与肿瘤转移 | CADM1、NME1、PI3K-AKT信号通路 | ||
| 参与血管形成 | RBMS3、PI3K-AKT信号通路 | ||
| 参与染色质重塑 | ATRX、ARID1A、ARID1B、EP300、CREBBP、KDM5A、IKZF1、CHD9 | ||
| 转录因子 | LMO1、WT1、PHOX2B、CAMTA1 | ||
| RNA结合蛋白 | LIN28、RBMS3 | ||
NB通常是在众多因素共同作用下发生的,如多个基因、一个基因的多个突变、基因的表观遗传学改变、环境作用及未知的随机因素。自2005年以来,GWAS常被用于肿瘤等研究,通过比较病例组与对照组之间SNP的差异频率来寻找风险变异因素。然而,目前的GWAS只涉及性状易感基因的很少一部分,能够解释的遗传度极大地低于预期值,并且极少数SNPs被明确具有与发病机制相关联的功能性作用。而且GWAS的结果往往存在假阳性、假阴性,检测到的单核苷酸多态性很少位于功能区以及对稀有变异和结构变异不敏感等问题,导致了其应用的局限性。基因芯片技术是应用已知核酸序列作为探针与互补的靶核苷酸序列杂交,由于不需要扩增,因此基因芯片是个相对封闭的系统,只能检测已知序列,缺乏发现新基因及基因改变的能力,而且应用基因芯片技术进行检测往往费用比较昂贵。新一代测序技术的进步,促进了全基因组测序、全基因组外显子测序以及靶基因深度测序的快速发展,也为解决上述问题提供了契机。外显子组序列仅占全基因组序列的1%,但大多数与疾病相关的变异位于外显子区,基因组定向捕获工具的出现也使全外显子组测序和靶基因深度测序成为可能[47]。因此,对靶基因进行捕获后的深度重测序是寻找NB潜在致病突变的关键方法,具有高度特异性和可行性,同时也为治疗干预措施提供潜在的新靶点。
国内外相关文献已报道了一些与NB发生及预后相关的基因改变,但随着近几年高通量测序技术的发展,通过对儿童恶性肿瘤发生相关的一系列基因进行靶向捕获后的深度重测序,可能发现与NB相关的未被明确的基因改变,并可进一步阐明NB在分子领域的发病机制,优化早期诊断及预后判断,以及发现潜在的治疗新靶点。
所有作者均声明不存在利益冲突

























