
长Q-T间期综合征(Long Q-T syndrome,LQTS)是一种单基因遗传性心脏离子通道病,以QT间期延长、T波异常、尖端扭转型室速(torsade de pointes,TdP)为心电图表现,反复发作晕厥、抽搐、甚至猝死为临床特征。尽管LQTS的总体患病率不高,但由于高发心源性猝死,已引起心血管医师的密切关注。目前国内缺少针对性的LQTS临床实践指南。本指南的编写参考了国内外本领域的基础研究、临床研究和其他国家的相关指南共识,对LQTS的临床表现、遗传学机制、诊断标准、治疗与预后、遗传咨询等方面进行总结,以期促进和规范其临床诊疗实践。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
长Q-T间期综合征(Long Q-T syndrome,LQTS)是一组常染色体单基因遗传病,由心肌细胞离子通道蛋白或其调控蛋白功能异常所致,以QT间期延长、T波异常、尖端扭转型室速(torsade de pointes,TdP)为心电图表现,反复发生晕厥、抽搐、甚至心源性猝死(sudden cardiac death,SCD)为临床特征。大约50%的突变携带者缺乏典型的临床表现。LQTS的患病率约为1/2500,平均发病年龄为14岁。未经治疗的LQTS患者SCD的年发生率为0.03%~0.90%[1],晕厥的年发生率约为5%,10年病死率可达50%[2]。本病无显著的种族或地域分布差异[3,4,5]。在我国,LQTS患者以女性多见,从婴幼儿至老年均可发病,但以年轻人为主。疾病的诱因和发作症状与国外报道类似。
LQTS的临床表现又分为心脏事件(恶性心律失常)和心电图异常。心脏事件主要为TdP以及由TdP蜕变的心室颤动,导致晕厥、抽搐、SCD。LQTS主要累及年轻人,其心脏结构一般正常,通常无前驱症状。从基因突变的角度,LQTS可分为诸多亚型,其中LQT1~3型最为常见,占总体的>90%[6]。晕厥和SCD通常发生在40岁之前,尤其是LQT2和LQT3。而LQT1患者40岁之后发生心脏事件仍不少见,其诱因通常为:LQT1:剧烈运动(跑步、游泳等)和情绪应激(恐惧、惊吓和生气等);LQT2:突然的声音刺激(如闹铃惊醒);LQT3:休息和心率慢;LQT4:运动和精神紧张;LQT7:低钾[7,8]。
心电图异常表现为Q-T间期延长,ST-T异常(T波形态多变,出现U波或TU融合)、TdP以及心室颤动。LQTS基因亚型与心电图特征存在一定的关联(表1)[9,10]。LQT1具有平滑、基底部较宽的T波;LQT2具有低振幅、带切迹/双峰T波;LQT3具有晚发尖锐/双相T波,ST段平直或斜形延长。各型LQTS的心电图形态存在一定的重叠。在同一家系中,可观察到T波形态的不均一性。即便是同一LQTS患者,心电图T波图形也可能多变,QT间期可暂时正常化。对于隐匿性LQT1和LQT2(静息下QT间期正常),运动试验或儿茶酚胺注射能够揭示出其特征性心电图异常。长短周期现象和T波电交替是预测发生TdP的敏感指标。
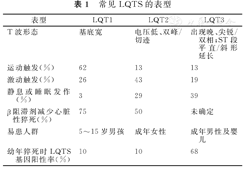
常见LQTS的表型
常见LQTS的表型
| 表型 | LQT1 | LQT2 | LQT3 |
|---|---|---|---|
| T波形态 | 基底宽 | 电压低、双峰/切迹 | 出现晚、尖锐/双相;ST段平直/斜形延长 |
| 运动触发(%) | 62 | 13 | 13 |
| 激动触发(%) | 26 | 43 | 19 |
| 静息或睡眠发作(%) | 3 | 29 | 39 |
| β阻滞剂减少心脏性猝死(%) | 75 | 50 | 未确定 |
| 易患人群 | 5~15岁男孩 | 成年女性 | 成年男性及婴儿 |
| 幼年猝死时LQTS基因阳性率(%) | 10 | 10 | 68 |
除心电图差异外,LQTS基因亚型还与其他临床特征存在关联:LQT7(Andersen-Tawil综合征)伴有肌肉无力、脊柱弯曲和面部畸形;LQT8(Timothy综合征)伴有皮肤畸形、神经发育异常和认知障碍;常染色体隐性遗传的Jervell-Lange-Nielsen综合征(Jervell and Lange-Nielsen syndrome,JLNS)伴有耳聋。
























