版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体病既往被认为是一种独立的疾病,但最近有临床报道却发现,MOG抗体可合并其他抗体共同致病,表现为并存疾病,其中MOG抗体合并抗N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)目前在国内尚少有成人病例报道。笔者现将广东三九脑科医院神经内科近年来收治的3例诊断为MOG抗体病合并抗NMDAR脑炎重叠综合征患者的临床诊治经过报道如下,以期为临床同道提供一些经验借鉴。
病例1 男性,51岁,因"反应迟钝伴幻觉2个月余"于2017年11月2日入院。体检:神志清晰,言语流利,反应迟钝,近期、远期记忆力下降,计算力减退,判断力、定向力正常,双眼视力下降,双侧瞳孔等大等圆、直径3 mm,直接及间接对光反射正常,余颅神经检查未见异常,四肢肌张力正常,肌力5级,共济运动检查未见异常,全身感觉系统检查未见异常,Romberg征(-),四肢腱反射正常,双侧病理征(-)。颈软,无抵抗。患者4个月前有双眼视力下降,当地医院考虑为视神经炎,予激素治疗,双眼视力逐渐恢复,遗留左眼视物不清。外院未行血清及脑脊液抗NMDAR抗体、MOG抗体、水通道蛋白4(aquaporin 4,AQP4)抗体、寡克隆区带检查。患者家族史、个人史无特殊,简易智力状态检查量表(MMSE)评分22分(初中学历)。腰穿脑脊液压力150 mmH2O(1 mmH2O=98 Pa),有核细胞数8×106/L,淋巴细胞为主,生化正常,血及脑脊液寡克隆区带、AQP4抗体均阴性,血清抗MOG-IgG阳性(1∶10),脑脊液抗MOG-IgG阴性,血清抗NMDAR抗体阳性(1∶100),脑脊液抗NMDAR抗体阳性(1∶10)。视诱发电位示双侧潜伏期延长,头颅MRI+增强扫描示双侧大脑半球皮层下白质内、双侧放射冠及半卵圆中心脱髓鞘病灶,增强未见异常强化(图1A、图1B)。全脊柱MRI+增强扫描未见明显异常。考虑诊断:MOG抗体病合并抗NMDAR脑炎重叠综合征。给予静脉滴注甲泼尼龙1000 mg 3 d,继而500 mg、240 mg、120 mg各3 d,然后改为口服泼尼松60 mg/d并逐渐减量,总疗程为半年。出院半年后电话随访,患者无幻觉、反应良好,遗留左侧视物稍欠清。
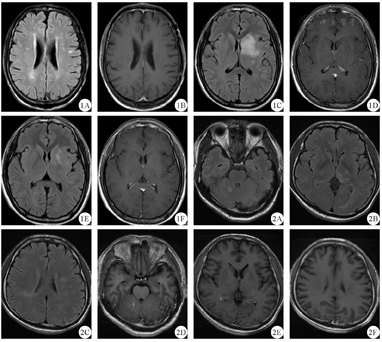
1A:病例1双侧放射冠及半卵圆中心FLAIR序列呈高信号,1B:病例1增强扫描后未见异常强化灶,1C:病例2左侧基底节区-放射冠-半卵圆中心FLAIR序列呈高信号,1D:例2软脑膜异常强化,1E:病例2 1个月后复查示病灶较前明显缩小,1F:病例2 1个月后复查示软脑膜异常强化减弱消失;2A~2C:病例3右侧小脑半球、双侧丘脑及双侧放射冠-半卵圆中心FLAIR序列呈高信号;2D~2F:病例3右侧小脑半球、双侧丘脑及双侧放射冠-半卵圆中心增强扫描后呈散在斑点状强化;FLAIR:液体衰减反转恢复
病例2 男性,16岁,因"头痛半个月,发热、右手无力、言语不能1周"于2020年3月13日入院。体检:神志清晰,言语欠流利,高级认知功能检查未见明显异常,12对颅神经检查未见明显异常,四肢肌张力正常,右侧肢体肌力4级,余肢体肌力5级,共济运动检查稳准,全身感觉系统检查未见异常,正常步态,四肢腱反射正常,双侧病理征(-)。颈软,无抵抗。患者既往史、家族史、个人史无特殊。腰穿脑脊液压力>330 mmH2O,有核细胞数94×106/L,生化正常,血及脑脊液寡克隆区带、AQP4抗体均阴性,血清及脑脊液抗MOG-IgG均阳性(1∶100),血清抗NMDAR抗体阴性,脑脊液抗NMDAR抗体阳性(1∶32)。视诱发电位示双侧潜伏期延长。头颅MRI+增强扫描示左侧基底节区-放射冠-半卵圆中心异常信号影,伴软脑膜异常强化,相应局部皮层稍肿胀(图1C、图1D)。全脊柱MRI+增强扫描未见明显异常。考虑诊断:MOG抗体病合并抗NMDAR脑炎重叠综合征。给予静脉注射丙种球蛋白400 mg/(kg·d) 5 d联合静脉滴注甲泼尼龙(1000 mg 5 d,500 mg 3 d,240 mg 2 d,120 mg 1 d),后改为口服泼尼松60 mg/d并逐渐减量,预计总疗程半年。2020年3月28日时患者言语流利,无头痛,右手无力出院。1个月后复查腰穿脑脊液压力225 mmH2O,有核细胞数8×106/L,头颅MRI+增强扫描可见病灶较前明显缩小(图1E、图1F),血及脑脊液寡克隆区带、AQP4抗体均阴性,血清抗MOG-IgG阳性(1∶1000),脑脊液抗MOG-IgG阳性(1∶32),血清及脑脊液抗NMDAR抗体均阴性。复诊时,患者未诉头痛、肢体无力等不适,予继续随访患者症状及病灶变化情况。
病例3 男性,37岁,因"记忆力下降20 d,加重伴视力下降2 d"于2019年12月5日入院。体检:神志清,精神可,近期、远期记忆力下降,计算力减退,判断力、定向力减退,双耳听力粗测下降,视物模糊,余颅神经检查未见异常,四肢肌张力正常,肌力5级,共济运动检查未见异常,全身感觉系统检查未见异常,Romberg征(-),四肢腱反射正常,双侧病理征(-)。颈软,无抵抗。患者既往史、家族史、个人史无特殊,MMSE量表评分19分(小学学历)。腰穿脑脊液压力155 mmH2O,有核细胞数16×106/L,淋巴细胞为主,生化正常,血及脑脊液寡克隆区带、AQP4抗体均阴性,血清抗MOG-IgG阳性(1∶10),脑脊液抗MOG-IgG阴性,血清抗NMDAR抗体阴性,脑脊液抗NMDAR抗体阳性(1∶32)。视诱发电位示左侧潜伏期延长,右侧正常。头颅MRI+增强扫描示右侧小脑半球、双侧丘脑及双侧半卵圆中心多发病灶,考虑脱髓鞘病变可能性,散在斑点状强化(图2)。考虑诊断:MOG抗体病合并抗NMDAR脑炎重叠综合征。给予静滴地塞米松磷酸钠10 mg/d,2 d后患者精神较亢奋,理解力较前下降,出现幻觉,遂改为静脉滴注甲泼尼龙1000 mg/d加用奥氮平2.5 mg/d。当甲泼尼龙1000 mg静滴5 d后减至500 mg静滴时,患者自觉视物不清较前加重,不配合治疗出院。出院后半年电话随访,患者仍有视物模糊,存在被害妄想,预后欠佳。
MOG抗体介导的MOG抗体病临床表现多样,与急性播散性脑脊髓炎、视神经脊髓炎谱系疾病、视神经炎和少部分多发性硬化均有重叠[1,2,3,4]。MOG抗体病诊断国际共识提出其诊断需符合以下条件:单发或复发性急性视神经炎、脊髓炎、脑干脑炎、脑炎或任何这些症状的并发,及MRI或视诱发电位提示中枢神经系统脱髓鞘相关病灶,以及血清MOG抗体阳性[5]。而抗NMDAR脑炎的特征性临床表现符合弥漫性脑炎,与经典的边缘性脑炎有所不同,常见头痛、发热等前驱症状,临床表现常包括精神行为异常、认知障碍、近事记忆力下降、癫痫发作、言语障碍、运动障碍、不自主运动、意识水平下降与昏迷、自主神经功能障碍等。通常来说,MOG抗体病及抗NMDAR脑炎均女性多见,但我们报道的这3例患者却均为男性,这可能与样本量小有关。其中,病例1及病例3均有视力下降的临床表现,且视诱发电位提示视神经受累,符合MOG抗体病的表现;病例1及病例3也均存在记忆力下降,MMSE量表评估结果进一步支持其存在认知障碍,同时病例1还存在幻觉以及病例3出院后随访时出现关系妄想等症状,这均符合抗NMDAR脑炎的临床表现。至于病例2,其出现了前驱感染,存在言语障碍、运动障碍,更符合抗NMDAR脑炎的临床表现,虽然其视力未见明显下降,但视诱发电位结果已提示视神经受累。总之,我们报道的这3例患者的临床表现均兼具MOG抗体病和抗NMDAR脑炎这两种疾病的临床表现。
近年来有研究报道部分抗NMDAR脑炎伴中枢神经系统脱髓鞘患者的血清MOG抗体阳性,以及部分MOG抗体病患者脑脊液抗NMDAR抗体阳性,即该类疾病兼具MOG抗体病和抗NMDAR脑炎这两种疾病的特点,故称之为MOG抗体病合并抗NMDAR脑炎重叠综合征[2,7]。我们报道的这3例患者均有MOG抗体病及抗NMDAR脑炎的临床表现,且血清MOG抗体、脑脊液抗NMDAR抗体均为阳性。
有研究报道MOG抗体病合并抗NMDAR脑炎重叠综合征在儿童中的发生率高于成人[8],但我们报道的这3例患者的年龄均在14岁以上,这可能与样本量小有关。
MOG抗体病合并抗NMDAR脑炎重叠综合征通常有精神行为异常、认知障碍等抗NMDAR脑炎症状,但这些症状多较经典的抗NMDAR脑炎患者轻[3,8,9]。我们报道的这3例患者中,病例1、病例3均有幻觉等精神行为异常,且病例1及病例2的抗NMDAR脑炎的症状较轻,与文献报道一致。
MOG抗体病患者的头颅MRI可见T2WI上高信号的脱髓鞘病灶成斑片状弥漫分布,边缘不清,大脑半球病灶可表现为大片状,也可见于皮层、放射冠、侧脑室周围和丘脑、基底核等深部核团,幕下病灶可累及大脑脚、桥脑、延髓、小脑半球,其脊髓病变常累及脊髓圆锥和胸、腰椎脊髓[10]。而抗NMDAR脑炎患者的头颅MRI可无明显异常,或仅有散在的皮质、皮质下点片状异常,少部分患者可有中枢神经系统脱髓鞘病变的影像改变,累及大脑白质和脑干等[6]。与MOG抗体病易出现脑干和脊髓病灶不同,MOG抗体病合并抗NMDAR脑炎重叠综合征患者均会出现幕上病灶,较少出现幕下及脊髓病灶[2,8]。我们报道的这3例患者中,病例1和病例2均只出现幕上病灶,病例3除了右侧小脑病灶外也以幕上病灶为主,且多位于皮层下白质。
目前有研究报道,在欧洲人种中,仅有约13%的MOG抗体病患者的寡克隆区带呈阳性,阳性率较低[5,11]。而我们报道的这3例患者的寡克隆区带均为阴性,这与上述报道符合的同时,或许也可能与样本量小有关。
抗NMDAR脑炎和MOG抗体病均需免疫治疗,一线免疫治疗效果不佳或复发病例需使用二线免疫治疗[5,6]。我们报道的这3例患者中,病例1及病例2予以一线免疫治疗后预后尚可,但病例3的效果欠佳且因其自行出院而未予二线免疫治疗,导致出院半年后患者仍有幻觉、视物模糊等症状。
MOG抗体病合并抗NMDAR脑炎重叠综合征的发生机制目前尚不明确,推测有以下一些可能:少突胶质细胞也可表达NMDAR。当针对髓鞘的异常免疫反应发生时,会同时累及NMDAR;并且MOG抗体病和抗NMDAR脑炎或许也可能存在共同的免疫途径异常[5,8]。具体的机制还需要以后更多的临床和基础研究来解答。
总之,在临床上,对疑诊中枢神经系统脱髓鞘病或抗NMDAR脑炎患者,我们建议应同时行自身免疫性脑炎相关抗体和中枢神经系统脱髓鞘相关抗体的检测,以协助明确诊断,同时制定最佳的治疗方案以减少复发及改善预后。
所有作者均声明不存在利益冲突


























