
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
红细胞增多症病因繁多,或由于组织缺氧引起的代偿机制,或由于JAK2酪氨酸激酶体细胞突变导致真性红细胞增多症(PV),或为遗传性红细胞增多症。遗传性红细胞增多症已知机制包括高氧亲和力血红蛋白变体(HOA)和二磷酸甘油酸变位酶(BPGM)缺乏所致血红蛋白病及红细胞生成素受体(EPOR)和氧敏感途径(OSP)异常引起的家族性红细胞增多症(familial erythrocytosis,FE)。其中OSP的最新研究已证实相关基因突变会导致von Hippel-Lindau(VHL,FE 2型)、脯氨酰羟化酶结构域2(PHD2,FE 3型)、低氧诱导因子2α亚基(HIF2A,FE 4型)功能异常继发FE[1,2]。我科近期收住一例VHL基因纯合突变致FE 2型患者,为国内首次报告,现对其基因突变特征及致病的分子机制、家系、临床资料等分析如下。
患者,男,21岁,主因"齿龈渗血半年,伴头痛头晕1个月"于2019年11月就诊,查血常规示:WBC 8.04×109/L,RBC 7.82×1012/L,HGB 272 g/L,PLT 85×109/L,以"红细胞增多症"收住我科。患者既往有红细胞增多病史3年余。入院查体:神清,颜面及颈部潮红,口唇、甲床呈紫红,周身浅表淋巴结未触及肿大,心肺腹查体未见异常,双下肢无水肿。末梢血涂片:成熟红细胞呈地毯样密集分布。血生化:葡萄糖(空腹)3.2 mmol/L(正常参考值3.9~6.1 mmol/L),尿酸478 μmol/L。凝血全套:纤维蛋白原1.8 g/L(正常参考值2.2~5.0 g/L),其他指标正常;红细胞生成素(EPO)浓度26.65 U/L(正常参考值2.59~18.50 U/L);肝肾功能、电解质、免疫球蛋白、心肌酶谱、肿瘤标志物、贫血三项(叶酸、维生素B12、血清铁蛋白)、淋巴细胞亚群、降钙素原、尿便常规均正常;类风湿因子、抗ENA、抗核抗体、血及尿免疫固定电泳均阴性;EB病毒(EBV DNA)定量<1×103。骨髓象:骨髓增生活跃,红系比例增高,未见早幼红细胞,中幼红细胞占0.120(正常参考值0.026~0.107),晚幼红细胞占0.230(正常参考值0.052~0.175)。骨髓活检:①骨髓有核细胞增生活跃,脂肪组织大致正常;②粒系增生,各阶段细胞比例正常;③红系增生异常活跃,以晚幼红为主;④巨核系增生减低,形态大致正常;⑤淋巴及浆细胞散在分布;⑥可见局灶少量的纤维组织。骨髓染色体核型分析未见异常;骨髓JAK2 V617F及12~15号外显子、MPL、CALR检测未见异常;心脏彩超正常;头胸腹增强CT未见异常。胃肠镜检查未见异常。患者父母非近亲结婚,家族史及遗传史无异常。本研究已征得患者及其父母知情同意,并获得本院伦理委员会批准。
基因检测:患者携带有VHL基因纯合突变c.598C>T(p.Arg200Trp),区带3p25.3,序列NM_000551.3,位置Exon3,为错义突变(图1A),患者上述突变分别遗传自其父母,c.598C>T的杂合核苷酸突变,家系验证来源于父亲,c.598C>T的杂合核苷酸突变,家系验证来源于母亲,其父母均只携带其中1个杂合突变,为正常表型(图1B、C)。VHL基因纯合突变在金域医学数据库KMTD、千人基因组数据库1000 genomics均未见收录。美国国家心肺血液研究所ESP6500数据库有收录(0.0002)。生物信息学软件预测其致病可能性大。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics,ACMG)2015指南,该突变分类为病理性突变。上述突变不属于多态性变化,在人群中发生的频率极低,上述突变可能导致蛋白质功能受到影响,结合患者临床表现、实验室检查、影像学检查,确诊为家族性红细胞增多症2型。
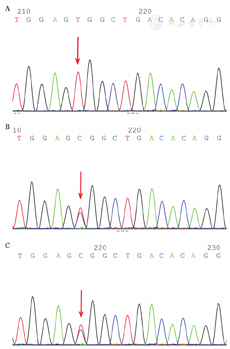
A:患者VHL 3p25.3 NM_000551.3 Exon3 c.598C>T p.(Arg200Trp)纯合突变;B:患者母亲VHL 3p25.3 NM_000551.3 Exon3 c.598C>T p.(Arg200Trp)杂合突变;C:患者父亲VHL 3p25.3 NM_000551.3 Exon3 c.598C>T p.(Arg200Trp)杂合突变
FE是常染色体隐性遗传的红细胞增多症,发病率极低,临床罕见。根据发病原因分为原发性和继发性,原发性为EPOR基因突变,为FE 1型,也称为先天性红细胞增多症(CE)或原发性先天性家族性红细胞增多症[3,4,5]。继发性FE是由于OSP相关基因突变,导致促红细胞生成物质(主要为EPO)调节异常,血清EPO水平升高[6],继发红细胞增多。
本例患者VHL等位基因发生错义突变并纯合突变,其父母均为杂合突变表型正常,为隐性遗传,且缺乏原发和继发红细胞增多的证据,EPO浓度增高,可明确诊断为FE 2型。FE 2型与VHL病鉴别如下:VHL病通常均以常染色体显性的方式遗传,VHL基因缺陷位于染色体3p25.5,如发生体细胞突变,还可引起小脑成血管细胞瘤、肾细胞癌;临床表现有红细胞增多症、神经系统血管母细胞瘤、嗜铬细胞瘤、肾细胞癌或肾囊肿、胰腺肿瘤或胰腺囊肿、内耳淋巴囊肿和生殖系统囊肿等,其临床表型多变[7]。FE 2型为常染色体隐性遗传,VHL基因缺陷位于染色体3p25-26,临床表现有红细胞增多、红细胞比容升高、血红蛋白升高、静脉曲张、头痛等。
随着对FE的深入研究,2002年Ang等[8]发现VHL基因突变,定位在3号染色体上,提出pVHL功能的破坏会导致HIF-1α降解失败,从而导致HIF-1α积累,下游靶基因(如EPO)上调以及红细胞增多症的临床表现。Mallik等[9]对FE研究发现纯合VHL c.598C>T(p.Arg200Trp)是FE最常见的致病突变,由此使FE的发病机制逐渐被阐明。VHL基因全长约为16 kb,包含E1、E2、E3三个外显子,可翻译成两种蛋白异构体,即含有213个氨基酸残基的pVHL30和含有160个氨基酸残基的pVHL19,这两种异构体生物学效应相似[10]。pVHL涉及多种功能,研究最多的是OSP,即HIF-PHD通路,其主要参与者是HIF,HIF是一种低氧依赖性的转录因子,它以3种亚型HIF-1α、HIF-2α和HIF-3α存在,通过激活基因转录参与调控机体对缺氧的全身和局部应答。在正常氧气供应下,HIF的α亚基被氧化,随后被PHD羟基化,然后与pVHL结合,pVHL是E3泛素连接酶复合物的一个亚基,能直接与转录延长因子Elongin C连接,再结合Elongin B、CUL2蛋白和环指蛋白RBX1最终形成VCB-CR复合体,该复合体属于E3泛素连接酶系统,可促进HIF-α泛素化并随后在蛋白酶体降解[11],从而不会产生EPO。PHD是HIF降解的限速酶,在缺氧或VHL基因突变情况下,PHD活性降低,使HIF-α亚基保持稳定,并与HIF-β亚基结合成二聚体,构成功能性HIF复合物易位至细胞核,并与靶基因的缺氧反应元件结合,转录激活与适应减少氧气供应有关的包括EPO在内的200多个基因的表达[12,13]。该患者VHL基因突变致缺氧途径的失调,EPO水平增高,是红细胞增多的根本原因。
多数FE 2型患者无症状,部分患者因红细胞增多引起血液流速减慢,血液淤滞,造成组织器官缺氧,引起一系列的临床症状,最常见是反复头痛头晕,其他表现包括疲劳、怕热、胸闷、胸痛、高血压、劳累性呼吸困难及腹痛和脾肿大,血栓并发症包括脑血管意外、短暂性脑缺血发作、肺栓塞、肢体血栓形成、门静脉血栓形成和深静脉血栓形成等[14]。该患者主要表现为头痛头晕同时伴有间断齿龈少量渗血,齿龈渗血考虑可能与原发病、血小板及纤维蛋白原减低等相关,具体原因仍不明确。实验室检查提示患者血糖(空腹)波动于3.0~3.4 mmol/L,考虑与FE相关。McClain等[15]报道VHL 598C> T纯合突变与较低的葡萄糖浓度和较低的糖基化血红蛋白水平相关,HIF-1a或HIF-2a上调可能导致血清葡萄糖浓度降低,葡萄糖转运蛋白GLUT2和糖异生葡萄糖6磷酸酶(G6PC)的肝表达降低,以及葡萄糖异生磷酸烯醇丙酮酸羧激酶的限速酶表达降低,但GLUT1或PDK2的表达没有明显增加。这些发现表明,肝糖原异生的损害导致了FE患者血糖的总体下降,导致葡萄糖从肝脏的释放受损,肝糖原储存增加和低血糖。
综上,我们报道了国内首例VHL基因c.598C>T纯合突变所致FE 2型病例,并探讨了FE 2型遗传学病因和发病的分子基础。

























