
探讨45,X/46,XY嵌合体患儿性腺特征、性腺肿瘤的发生率及SRY基因及Y染色体微缺失检测结果。
回顾性分析2013年1月至2019年12月在江西省儿童医院就诊的45,X/46,XY核型或其变异型患儿的病例资料,对45,X/46,XY嵌合体性腺表型及分子生物学进行分析。
30例45,X/46,XY核型或其变异型患儿中,就诊年龄均在18岁以下,社会性别男性11例,社会性别女性19例,所有患儿中14例已行预防性性腺切除术。单侧睾丸和对侧条纹性腺检出6例,社会性别均为男性,病理切片显示性腺组织中同时含睾丸和卵巢组织者3例;同时伴有易位的肾上腺组织2例;双侧条纹性腺检出8例,社会性别均为女性,病理切片显示性腺组织中同时含附睾和卵巢组织者1例,性腺母细胞瘤1例,卵巢发育不良伴粒层细胞瘤样增生1例,有增生痣细胞(混合痣)1例。所有B超检查及病理切片均未发现卵泡存在。11例社会性别男性患儿中,5例经SRY基因检测结果均阳性,7例经Y染色体微缺失检测,显示Y染色体部分缺失3例,无缺失4例;19例社会性别女性患儿中,10例经SRY基因检测,其中9例结果阳性,1例阴性;7例经Y染色体微缺失检测,显示Y染色体部分缺失2例,无缺失4例,全部缺失1例。
45,X/46,XY嵌合体患儿大多含有异常的性腺组织,具有发生性腺肿瘤的风险,尤其女性患儿发病率较高,绝大部分患儿SRY基因阳性,Y染色体无缺失或部分缺失。考虑到此类患儿发生性腺肿瘤的风险增加,建议早期进行预防性性腺切除术。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
在性发育障碍中,45,X/46,XY嵌合体是罕见的,估计发病率为1/10 000新生儿。据推测[1,2,3],45,X/46,XY核型的产生,可能是由于45,X细胞系在胚胎有丝分裂早期,一个正常或结构异常的Y染色体丢失,从而产生镶嵌现象,其临床表型差异很大,从具有特纳综合征特征的女性表型到生殖器性腺模糊的性发育异常表型,再到完全男性化表型,均有分布。性腺形状及病理组织形态也差异较大,发生性腺肿瘤的风险不一,Y染色体性别决定区(sex-determining region of Y-chromosome,SRY)基因检测及Y染色体微缺失检测在发现Y染色体片段有一定的参考价值。目前,关于45,X/46,XY核型及其变异核型的研究较少,尤其是针对儿童青少年患者的研究更为少见。本研究旨在探讨45,X/46,XY核型患儿的性腺特征、性腺肿瘤的发生率、SRY基因及Y染色体微缺失检测结果,并结合相关文献对研究结果进行分析。
收集2013年1月至2019年12月在江西省儿童医院就诊,经染色体核型分析确诊为45,X/46,XY核型或其变异核型的患儿30例,社会性别男性11例,社会性别女性19例,年龄1~16岁。本研究通过医院医学伦理委员会批准(批准文号:JXSETYY-YXKY-20200041),研究对象监护人均知情同意,并签署知情同意书。
取外周血淋巴细胞培养制备染色体,G显带技术分析核型,油镜下每例计数中期分裂象,至少计数30个核型。异常核型根据TSCN1995人类细胞遗传学国际命名体制进行命名,染色体核型为45,X/46,XY及其变异核型的患儿纳入研究。
排除染色体核型虽符合入选标准,但既无具体的性腺表型资料,包括性腺外观、性腺术中探查表现及性腺病理检查结果,也未进行外周血SRY基因及Y染色体微缺失检测的患儿。
收集患儿临床资料,包括具体染色体核型,术前性腺部位彩超检查结果,术中性腺形态,术后性腺组织病理切片分析结果,外周血SRY基因与Y染色体微缺失检测结果。取患儿肘静脉血,采用TIANGEN公司(中国,北京)提供的血液基因组DNA提取试剂盒提取外周血DNA,设计SRY基因引物进行PCR扩增,进行琼脂糖凝胶电泳。采用上海透景生命科技公司提供的Y染色体微缺失检测试剂盒进行Y染色体微缺失检测。操作步骤严格按照试剂盒说明书进行。
采用计数资料方法进行描述。
本组纳入的30例患儿中,染色体核型为标准45,X/46,XY核型者26例;45,X/47,XXY核型1例;45,X/47,XYY核型1例;45,X,der(18,Y)核型1例;45,X/46,XY/46,X,del(Y)核型1例。
本组纳入45,X/46,XY核型及其变异核型患儿30例,其中社会性别男性11例,社会性别女性19例,就诊年龄均在18周岁以下。22例患儿在术前经子宫卵巢或阴囊腹股沟部位的B超检查,提示双侧性腺显示不清者7例,提示一侧性腺显示不清或不可及者6例,B超可探及双侧性腺者9例。
女性表型患儿中,5例有发际线低,乳间距宽等Turner综合征外貌,1例有先天性心脏病室间隔缺损,1例桥本甲状腺炎伴脊柱侧凸,2例肾盂分离,1例先天性髋关节脱位,1例垂体Rathke囊肿。
所有患儿中,15例行促性腺激素释放激素(GnRH)激发试验,生长激素(GH)峰值>7 μg/L 6例,GH峰值<7 μg/L 9例,此15例患儿身高均低于儿童青少年标准身高的-2s。
14例已手术患儿的具体染色体核型及其嵌合比例、性腺表型、病理结果见表1。此14例患儿已选择行预防性性腺切除术,其中6例社会性别男性,8例社会性别女性。
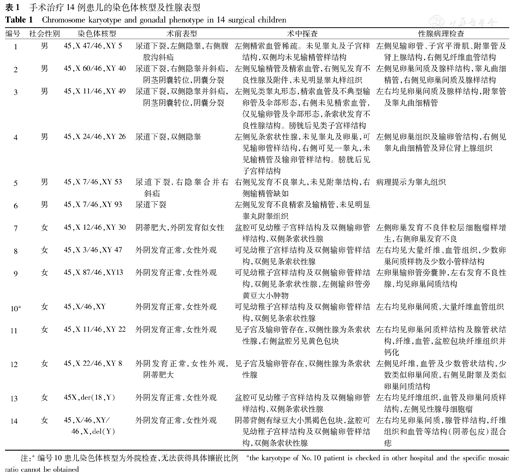
手术治疗14例患儿的染色体核型及性腺表型
Chromosome karyotype and gonadal phenotype in 14 surgical children
手术治疗14例患儿的染色体核型及性腺表型
Chromosome karyotype and gonadal phenotype in 14 surgical children
| 编号 | 社会性别 | 染色体核型 | 术前表型 | 术中探查 | 性腺病理检查 |
|---|---|---|---|---|---|
| 1 | 男 | 45,X 47/46,XY 5 | 尿道下裂,左侧隐睾,右侧腹股沟斜疝 | 左侧精索血管稀疏。未见睾丸及子宫样结构,双侧均未见输精管样结构 | 左侧见输卵管、子宫平滑肌、附睾管及肾上腺结构,右侧见纤维血管结构 |
| 2 | 男 | 45,X 60/46,XY 40 | 尿道下裂,右侧隐睾并斜疝,阴茎阴囊转位,阴囊分裂 | 左侧见输精管及精索血管,右侧见发育不良性腺及附件,未见明显睾丸样组织 | 左侧见卵巢间质及腺样结构,睾丸曲细精管,右侧见卵巢间质及腺样结构 |
| 3 | 男 | 45,X 11/46,XY 49 | 尿道下裂,双侧隐睾并斜疝,阴茎阴囊转位,阴囊分裂 | 左侧见类睾丸形态,精索血管及不典型输卵管及伞部形态,右侧未见精索血管,仅见输卵管及伞部形态,条索状发育不良性腺结构。膀胱后见类子宫样结构 | 左右均见卵巢间质及腺样结构,附睾管及睾丸曲细精管 |
| 4 | 男 | 45,X 24/46,XY 26 | 尿道下裂,双侧隐睾 | 左侧见条索状性腺,未见睾丸及卵巢,可见输卵管样结构,右侧可见一睾丸,未见输精管及输卵管样结构。膀胱后见子宫样结构 | 左侧见卵巢组织及输卵管结构,右侧见睾丸曲细精管及异位肾上腺组织 |
| 5 | 男 | 45,X 7/46,XY 53 | 尿道下裂,右隐睾合并右斜疝 | 右侧见发育不良睾丸,未见附睾结构,右侧输精管缺如 | 病理提示为睾丸组织 |
| 6 | 男 | 45,X 7/46,XY 93 | 尿道下裂 | 左侧见发育不良精索及输精管,未见明显睾丸附睾组织 | |
| 7 | 女 | 45,X 12/46,XY 30 | 阴蒂肥大,外阴发育似女性 | 盆腔可见幼稚子宫样结构及双侧输卵管样结构,双侧条索状性腺 | 左侧卵巢发育不良伴粒层细胞瘤样增生,右侧卵巢发育不良 |
| 8 | 女 | 45,X 3/46,XY 47 | 外阴发育正常,女性外观 | 可见幼稚子宫样结构及双侧输卵管样结构,双侧见条索状性腺 | 左右均见大量纤维、血管组织,少数卵巢间质样物及少数小管样结构 |
| 9 | 女 | 45,X 87/46,XY13 | 外阴发育正常,女性外观 | 可见幼稚子宫样结构及双侧输卵管样结构,双侧见条索状性腺,左侧输卵管旁黄豆大小肿物 | 左卵巢输卵管旁囊肿,左右发育不良性腺,均见卵巢间质结构 |
| 10a | 女 | 45,X/46,XY | 外阴发育正常,女性外观 | 可见幼稚子宫样结构及双侧输卵管样结构,双侧见条索状性腺 | 左右均见卵巢间质,大量纤维血管组织 |
| 11 | 女 | 45,X 11/46,XY 22 | 外阴发育正常,女性外观 | 见子宫及输卵管存在,双侧性腺为条索状性腺,右侧盆腔另见黄色包块 | 左右均见卵巢间质样结构及腺管状结构,纤维,血管,盆腔包块纤维组织并钙化 |
| 12 | 女 | 45,X 22/46,XY 8 | 外阴发育正常,女性外观,阴蒂肥大 | 见子宫及输卵管存在,双侧性腺为条索状性腺 | 左侧见纤维,血管及少数管状结构,少数类似卵巢间质,右侧见附睾及类似卵巢间质结构 |
| 13 | 女 | 45X,der(18,Y) | 外阴发育正常,女性外观 | 盆腔可见幼稚子宫样结构及双侧输卵管样结构,双侧条索状性腺 | 左右均见纤维组织,血管及卵巢间质样结构,左侧见性腺母细胞瘤 |
| 14 | 女 | 45,X/46,XY/46,X,del(Y) | 外阴发育正常,女性外观 | 阴蒂背侧有绿豆大小黑褐色包块,盆腔可见幼稚子宫样结构及双侧输卵管样结构,双侧条索状性腺 | 左右均见卵巢间质,腺管样结构,纤维组织和血管等结构(阴蒂包皮)混合痣 |
注:a编号10患儿染色体核型为外院检查,无法获得具体镶嵌比例 athe karyotype of No.10 patient is checked in other hospital and the specific mosaic ratio cannot be obtained
6例男性表型患儿行腹腔探查术,术中发现单侧睾丸和对侧条索状性腺2例,单侧睾丸和对侧发育不良性腺4例,病理切片显示性腺组织中同时含睾丸组织和卵巢间质者3例,此3例男性表型患儿中2例的病理组织中同时伴有易位的肾上腺组织,术后病理检查未发现性腺肿瘤的发生;所有男性表型患儿均有尿道下裂,5例合并隐睾,4例合并斜疝。
8例女性表型患儿行腹腔探查术,术中发现7例均有双侧条索状性腺,病理切片显示性腺组织中同时含附睾和卵巢间质者1例,发生性腺母细胞瘤1例,卵巢发育不良伴粒层细胞瘤样增生1例,性腺真皮和表皮内见增生的痣细胞(混合痣)1例。所有B超检查及病理切片均未发现卵泡的存在。
本研究中,男性表型患儿尚未发现性腺肿瘤或癌前病变的病例,女性患儿中已发生性腺肿瘤1例,瘤样增生及痣细胞增生各1例。45,X/46,XY核型及其变异核型的女性患儿性腺肿瘤的发生风险高于男性患儿。含Y染色体的细胞嵌合比例高的患儿共6例,男性、女性表型各3例,且此6例患儿性腺只有睾丸组织,或只有卵巢组织,或同时有睾丸和卵巢组织,45,X/46,XY核型嵌合比例和性腺表型并无明显相关性。
11例社会性别男性患儿中,5例经SRY基因检测结果均阳性,7例经Y染色体微缺失检测,显示Y染色体部分缺失者3例,无缺失者4例;19例社会性别女性患儿中,10例经SRY基因检测,结果均阳性;7例经Y染色体微缺失检测,显示Y染色体部分缺失者2例,无缺失者4例,全部缺失者1例。由此看出,15例45,X/46,XY核型及其变异核型的患儿经SRY基因检测,均阳性;14例经Y染色体微缺失检测,全部缺失者仅有1例,其余均为部分缺失或全缺失。说明SRY基因及Y染色体微缺失检测在分子水平上发现Y染色体有一定的参考价值,可与细胞水平发现Y染色体互相印证。
45,X/46,XY核型包括Y染色体的结构畸变,如缺失、等双着丝粒等,异常的Y染色体可能导致其在部分细胞中的丢失[1,2,3]。一侧是条索状性腺,对侧是睾丸为混合性性腺发育不全(mixed gonadal dysgenesis,MGD);两侧都是条索状组织为完全性腺发育不全(complete gonadal dysgenesis,CGD);睾丸发育不良为部分性腺发育不全(partial gonadal dysgenesis,PGD)[4]。本研究中,14例行预防性性腺切除术,6例男性表型患儿术中发现单侧睾丸和对侧条索状性腺2例,单侧睾丸和对侧发育不良睾丸4例,8例女性患儿经腹腔探查术,术中发现7例为双侧条索状性腺,1例为双侧发育不良卵巢组织。因此,本组14例行预防性性腺切除术的嵌合体中,MGD共2例,PGD共5例,CGD共7例。据Matsumoto等[5]报道,34例45,X/46,XY嵌合体中,其中19例男性表型,男性表型所占比例更大,单侧睾丸和对侧条纹性腺检出20例(59%),双侧条纹性腺检出9例(26%),双侧睾丸检出5例(15%),结果和本研究结果有所差异,可能是因为男性表型与女性表型患者所占比例差异较大。本研究发现22例患儿的B超结果中有9例可探及双侧性腺,而这22例中的14例行预防性性腺切除术,仅有5例双侧均为发育不良性腺,其余均至少有一侧为条索状组织。因此,即使B超探查可及双侧性腺,也不能排除性腺发育不良或条索组织的存在。Ljubicic等[6]在所有研究的性腺组织中均未检测到卵泡,推测可能是因为在原始卵泡形成之前卵原细胞已丢失,这一发现与之前关于45,X/46,XY核型性腺功能的研究结果[7]一致。本研究中术前B超检查及术后病理组织切片中也均未发现卵泡存在。
有研究发现45,X/46,XY嵌合体程度与生殖器表型之间并无明显相关性[8],但这项研究的局限性在于研究过程中是用外周血进行检测,并不一定反映性腺组织的真实镶嵌程度。最近的一项研究确定了不同程度的45,X/46,XY镶嵌比值,并发现45,X/46,XY比值与Turner综合征样表型呈正相关[9]。Hornig等[10]发现雄激素受体活性与外部男性化程度呈正相关,雄激素受体表达和活性可能影响45,X/46,XY嵌合体患者的表型变异性。
本组6例男性表型患儿中,术后病理检查均未发现性腺肿瘤的发生;8例女性表型患儿中,发生性腺母细胞瘤1例,卵巢发育不良伴粒层细胞瘤样增生1例,性腺真皮和表皮内见增生的痣细胞,即混合痣1例。本组性腺肿瘤患儿表型均为女性,且性腺均为条索状组织。最近的研究报道,女性表型患者的风险最高[11,12,13],Cools等[14]报道,45,X/46,XY嵌合体患者的肿瘤风险在未成熟和/或低分化的性腺组织中最为明显,组织活检往往可能无法代表整个性腺的状况。45,X/46,XY嵌合体患者的性腺肿瘤总患病率为18%,腹腔内性腺的风险最高。考虑到性腺母细胞瘤的早期发生及其恶化的可能,建议早期行预防性性腺切除术,对尚保留睾丸组织的男性表型患者建议密切终身随访,且需要多学科联合诊治[15,16]。大多数确诊的患者在成年时因不育就诊[17,18]。Dumeige等[2]研究46例出生前与正常男性生殖器官发育异常相似的嵌合体患者,并随访其预后,发现大多数患者在成年时被诊断为无精子症。Y染色体结构缺陷主要是等臂Y色体Y(p),因为无精子因子(AZF)区域的缺失而影响生育能力[19]。然而,临床表型与45,X细胞的所占百分比无关,而是由45,X的细胞分布范围决定[20]。
本研究中1例有先天性心脏病室间隔缺损,1例桥本甲状腺炎伴脊柱侧凸,2例肾盂分离,1例先天性髋关节脱位,1例垂体Rathke囊肿。Klásková等[21]建议,不管这些患者最初的临床表现如何,均应进行心血管等重要器官检查。
本组15例嵌合体者行GnRH激发试验,GH峰值>7 μg/L 6例,GH峰值<7 μg/L 9例,此15例患儿身高均低于儿童青少年标准身高的-2s。Pan等[22]研究发现27例嵌合体中,17例出现身材矮小(低于-2 s)。最近的一项研究报告显示[23],XY mosaic Turner患者对生长的影响小于经典Turner女童。
本组15例患儿经SRY基因检测,除1例女性表型患儿结果阴性,其余结果均为阳性,包括9例女性表型患儿;14例经Y染色体微缺失检测,无缺失者8例,部分缺失者5例,男性、女性表型中均有发现,全缺失者1例,为女性表型患儿。Wu等[7]检测15例45,X/46,XY嵌合或其变异核型患者的Y染色体微缺失,发现6例存在Y染色体片段的缺失。Y染色体的结构重排,包括缺失、环状染色体和等色染色体,可能导致不同的表型。在回文组成的区域内有一个断点,这可能是这些缺失出现的原因[24,25]。包括SRY基因在内的短臂缺失直接影响睾丸分化,导致条纹性腺和女性表型,而长臂缺失,特别是涉及Yq11上AZFa、AZFb和AZFc的无精子症因子区域,导致男性不育[26]。AZFc最常参与其中,其次为AZFb,更少见的为AZFa。如果有Y微缺失,通过辅助生殖技术,可能产生不育的雄性后代,或者携带不稳定Y染色体的个体[27]。
综上所述,45,X/46,XY嵌合体患儿大多含有异常的性腺组织,具有发生性腺肿瘤的风险,尤其女性患儿发病率较高,绝大部分患儿SRY基因阳性,Y染色体无缺失或部分缺失。考虑到此类患儿发生性腺肿瘤的风险增加,建议早期进行预防性性腺切除术。
所有作者均声明不存在利益冲突

























