
探讨NCF2基因复合杂合突变导致的儿童慢性肉芽肿病(CGD)突变类型与临床特征、预后之间的关系。
分析2019年8月天津市儿童医院诊治的1例由NCF2基因复合杂合突变导致新生儿CGD的临床资料,并检索国内外文献,总结NCF2基因突变引起的CGD的临床特征、基因突变类型及预后等特点。
该患儿NCF2基因上存在复合杂合突变c.196_197insA(p.Arg66Glnfs23X)和c.1180T>G(p.Tyr394Asp),结合出生25 d发热、咳嗽、胸CT肺部多发团块影、结节影的临床表现、中性粒细胞呼吸爆发试验刺激指数23,确诊CGD。此突变在人类基因突变数据库中未见报道,为新突变。该患儿残留一部分烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶活性,服用抗真菌药物随访至9月龄未出现反复感染。国内外数据库报道NCF2基因突变的CGD患者共101例。有中性粒细胞呼吸爆发试验刺激指数结果者共33例,3以下22例;3以上11例,其中含错义突变8例。
c.196_197insA和c.1180T>G是NCF2基因的新突变,可导致CGD。含NCF2基因错义突变的CGD患者可能残留更多NADPH氧化酶活性。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
慢性肉芽肿病(CGD)是一种罕见的呼吸爆发缺陷引起的原发性吞噬细胞免疫缺陷病。该病由基因突变引起吞噬细胞烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶缺陷,导致NADPH氧化酶活性降低,不能产生活性氧(ROS)而致病。NCF2基因突变引起的CGD仅占所有CGD患者的5%,临床表现从症状轻微到严重结局,变异性大。本研究对天津市儿童医院呼吸科2019年8月诊治的1例NCF2基因突变导致的CGD患儿的临床特征、治疗和预后进行回顾,查阅文献,探讨NCF2基因突变的CGD临床特征与基因突变类型、预后的关系,提高医务工作者对儿童CGD的认识。
患儿,男,出生25 d起病,因"间断发热13 d伴咳嗽"入院。发热,最高体温38.7 ℃。咳嗽,偶有咳后面红,无喘息。大便略稀,6~8次/d,无腹胀、排尿哭闹,吃奶水量略减少。外院抗感染治疗仍间断发热,为进一步诊治转入天津市儿童医院。
个人史、家族史:患儿为第2胎,第2产,足月顺产,出生体质量4.1 kg,无窒息史。入院体质量4.7 kg,母乳喂养,出生21 d脐带脱落。接种卡介苗、乙肝疫苗第1针。无鸽粪、家禽、结核患者等接触史。母亲孕中期有念珠菌性阴道炎,外用药物治疗后好转。父母体健,非近亲结婚,否认家族遗传病及亲属幼儿夭折或流产史。有一姐姐,2岁,体健。
体格检查:神志清,反应可,呼吸促,60次/min,三凹征(-),无发绀,血氧饱和度98%。无皮疹及肛周破溃或脓肿,卡介苗接种处结痂,无红肿。双肺呼吸音粗,未闻及啰音,心腹及神经系统查体未见异常。
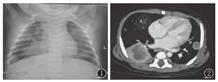
实验室检查:血常规示白细胞为(17.18~27.00)×109/L,血红蛋白、血小板正常,C反应蛋白最高135 mg/L;肝肾功能正常,降钙素原0.42 μg/L;血免疫球蛋白IgM 、IgG 、IgA、IgE正常;流式淋巴细胞亚群分类均正常;铁蛋白1 111 μg/L。血巨细胞病毒抗体阴性,1,3-β-D葡聚糖(G试验)阴性,血清半乳甘露聚糖(GM试验)2.13 μg/L(正常范围0~0.85 μg/L),γ-干扰素释放试验、血培养阴性。痰抗酸染色、真菌荧光染色、痰培养均阴性。胸片示双肺多发片状高密度影(图1)。胸CT平扫及强化示双肺多发软组织密度团块影及结节影伴边缘强化,纵隔内、右侧肺门及双侧腋窝多发淋巴结影并部分增大(图2)。胸部彩超示右侧胸腔实性包块,范围7 mm×26 mm×23 mm,考虑肺脓肿。
注:CGD:慢性肉芽肿病 CGD:chronic granulomatous disease
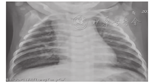
诊治经过:入院诊断重症肺炎、肺脓肿,予利奈唑胺联合头孢哌酮钠舒巴坦钠抗感染。入院第3天,结合患儿发热、咳嗽,肺部影像学特征性表现,血清GM试验单次>1.5 μg/L,临床诊断儿童侵袭性肺曲霉菌病[1]。予伏立康唑[负荷量9 mg/(kg·次),2次/d,1 d,改维持量8 mg/(kg·次),2次/d,静脉滴注],人免疫球蛋白(1 g/kg)。住院11 d,病情好转出院,继续口服伏立康唑[9 mg/(kg·次),2次/d]。出院后有头皮及面部的皮肤毛囊炎性丘疹。发病35 d复查G试验阳性,血清GM试验0.85 μg/L,铁蛋白降至737 μg/L。发病75 d复查胸片双肺多发片状高密度影明显吸收(图3)。服用伏立康唑随访至9月龄,未再合并感染,体质量增长至8.5 kg,生长发育适龄。
采用二氢罗丹明流式细胞分析法,测定佛波酯(PMA)刺激前后中性粒细胞内的ROS氧化二氢罗丹明产生的荧光强度并计算刺激指数(SI),进行中性粒细胞呼吸爆发功能筛查。用SI来评估NADPH氧化酶活性和ROS的产生能力,正常人群中性粒细胞SI的平均数为127.9(85.2~264.4)[2]。本患儿SI为23,提示中性粒细胞呼吸爆发功能下降。
经监护人知情同意,抽取患儿及父母静脉血各2 mL,行全外显子组的基因测序及Sanger测序进行验证。结果示患儿NCF2基因(NM_000433.3)存在2个位点的复合杂合突变,2号外显子存在c.196_197insA(p.Arg66Glnfs23X)移码突变,导致蛋白质66位精氨酸变为谷氨酰胺,并在后面23个氨基酸出现了终止密码子,导致截短蛋白;13号外显子存在c.1180T>G(p.Tyr394Asp)错义突变,导致蛋白质394位酪氨酸变为天冬氨酸。患儿母亲携带13号外显子c.1180T>G杂合错义突变,父亲携带2号外显子c.196_197insA杂合移码突变,患儿2个NCF2基因突变为复合杂合突变,符合常染色体隐性遗传规律(图4)。
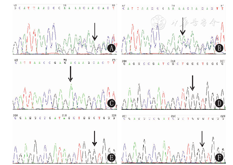
根据2017年"遗传变异分类标准与指南"[3]行致病性分析。c.196_197insA突变来源于父亲,使蛋白质翻译提前终止,预计会致所编码的蛋白质截短从而丧失正常功能(非常强致病性证据,PVS1);c.196_197insA突变通过比照千人基因组数据库、人类基因突变数据库、ESP6500siv2_ALL、dbSNP147数据库均未见收录(中等致病性证据,PM2);患儿临床表现与NCF2基因突变导致的CGD表型高度吻合(支持致病性证据,PP4)。c.196_197insA突变的证据强度为"PVS1+ PM2+PP4",判断为致病性突变。c.1180T>G来源于母亲,千人基因组数据库、人类基因突变数据库、ESP6500siv2_ALL未见收录(中等致病性证据,PM2);在隐性遗传病中,在反式位置上检测到致病变异(中等致病性证据,PM3);多种生物信息学软件预测其可能影响剪接,软件预测有害(支持致病性证据,PP3);患儿临床表现与CGD表型高度吻合(支持致病性证据,PP4)。c.1180T>G突变的证据强度为"PM2+PM3+PP3+PP4",判断为可疑致病性突变。
以"慢性肉芽肿病"检索中国知网、万方医学网,"chronic granulomatous disease"、"NCF2"检索PubMed,并未发现有关于c.196_197insA和c.1180T>G复合杂合突变的文献报道,上述突变为NCF2基因的新突变。结合患儿临床表现、中性粒细胞呼吸爆发试验、基因测序结果及致病性分析,确诊为CGD。
本研究通过医院医学伦理委员会批准(批准文号:L2020-20)。
以"慢性肉芽肿病"为关键词检索中国知网、万方医学网,以"chronic granulomatous disease"、"NCF2"为关键词检索PubMed自建库至2020年3月收录的文献,报道的NCF2基因突变CGD患者共101例。NCF2基因突变类型有错义突变、缺失、无义突变、插入突变及剪接突变等,其中错义突变48例(47.5%),占比最高。SI结果有记录的共33例,3以下22例;3以上11例,其中含错义突变8例。1岁内起病37例,1岁以上起病19例。近亲结婚31例(30.7%),非近亲结婚32例(31.6%),未描述38例(37.7%)。主要临床表现有肺炎、肺脓肿30例,淋巴结炎17例,腹泻15例,肝脾大、肝脾脓肿13例,皮肤脓肿7例,肛周脓肿3例,腹股沟、乳腺脓肿2例,卡介菌病12例、骨髓炎5例,肺结核4例、脑膜炎2例,炎症性肠病(inflammatory bowel disease,IBD)5例,自身免疫性疾病3例,脉络膜视网膜病变2例等,未描述临床表现39例。存活27例(26.7%),死亡25例(24.8%),预后未描述49例(48.5%)。
NADPH氧化酶由CYBB、CYBA、NCF1、NCF2和NCF4基因编码的5个亚基组成。NCF2基因突变导致的CGD为常染色体隐性遗传CGD,发病率低,全球共报道101例,30.7%的病例父母为近亲结婚,伊朗、以色列、土耳其等中东国家报道较多。
活化的NADPH氧化酶是参与呼吸爆发的关键酶,CGD患者的吞噬细胞不能进行正常的呼吸爆发产生ROS,从而使吞噬细胞不能杀伤所吞噬的过氧化物酶阳性的细菌与真菌,引起反复严重感染,产生过度炎症反应形成肉芽肿等CGD的多种临床表现。
多数CGD患者起病于感染,常初诊于呼吸科或消化科。本例患儿新生儿期起病于侵袭性曲霉菌感染引起的发热,进展为肺脓肿,合并皮肤毛囊炎性丘疹,临床主要表现为发热,肺部影像学为特征性的多发团块影、结节影伴边缘强化。与文献报道的X-连锁隐性CGD临床表现相似[4],肺炎最常见,其他依次为淋巴结炎、腹泻、脓肿、骨髓炎、肺结核等,还有IBD、自身免疫性疾病等。极早发炎症性肠病(very early onset IBD,VEO-IBD)有一部分可归为原发性免疫缺陷病[5]。文献复习101例NCF2基因突变的CGD表现有IBD者5例[6,7,8],均在出生6个月内出现慢性腹泻或便血,在出现反复严重感染之前,易被误诊为牛奶蛋白过敏或因合并自身免疫性疾病而诊断延迟,对VEO-IBD尤其2岁以内起病的患者,应进行CGD和其他免疫缺陷病的评估。
除上述临床表现,还可以通过吞噬细胞呼吸爆发功能、NADPH氧化酶组分蛋白的表达水平以及基因突变分析进行CGD的精准分子诊断[9]。有研究表明,SI水平与CGD发病年龄呈正相关[10]。ROS包括超氧阴离子及其他ROS中间产物(reactive oxygen intermediates,ROI),相比无ROI的CGD患者,残留一部分ROI的患者发病年龄晚,临床症状相对轻微,严重感染率低,存活时间较长[11,12]。本患儿SI为23,提示残留一部分产生ROS的能力,因此除了曲霉菌感染引起肺脓肿,随访患儿至9月龄,目前无反复感染及自身免疫性疾病,远期预后还有待观察。文献报道的101例CGD死亡例数占24.8%,有SI结果的33例中,3以下22例、3以上11例,可见NCF2基因突变的CGD残留NADPH氧化酶活性差异较大,临床表现和预后的变异性也大[12]。
NCF2基因突变类型多样,错义突变发生率最高,有SI结果3以上11例中含错义突变8例,提示含NCF2基因错义突变的CGD患者可能残留有更多NADPH氧化酶活性,临床病情轻,预后较好[11]。本例患儿为错义突变合并移码突变,也残留一部分产生ROS的能力,在及时诊断及积极的抗真菌药物治疗和预防下,目前生长发育适龄。但即使有相同的NCF2基因突变,临床表现从症状轻微到严重结局也会不同,除与NADPH酶活性相关外,环境条件和医疗护理等因素也会影响疾病进程[10]。
本例患儿NCF2基因存在c.196_197insA和c.1180T>G复合杂合突变,丰富了NCF2基因的突变谱系,为CGD的分子诊断提供新的依据。对CGD的临床表现、中性粒细胞呼吸爆发功能检查及基因筛查等精准分子诊断,可以帮助尽早识别CGD,尽早应用抗真菌药或抗生素预防感染、γ-干扰素治疗,积极创造条件进行造血干细胞移植来改善预后。
所有作者均声明不存在利益冲突

























