
法布雷病是一种罕见的X连锁遗传性溶酶体脂质贮积病,是由于GLA基因突变导致α-半乳糖酶A(α-Gal A)活性下降或缺失,引起鞘糖脂代谢紊乱,造成三己糖酰基鞘脂醇(GL-3)及其衍生物脱乙酰基GL-3(Lyso-GL-3)等糖鞘脂在组织中进行性累积,最终导致多器官系统病变。临床表现缺乏特异性,需结合酶活性、生物标志物GL-3和Lyso-GL-3的测定、病理及基因检测明确诊断。目前的治疗方法主要是酶替代疗法和口服药物伴侣。现就法布雷病的诊断和治疗进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
法布雷病(Fabry disease),又称"Anderson-Fabry病"(Anderson-Fabry disease,AFD),是一种X连锁遗传性溶酶体贮积症(LSD),因位于X染色体长臂q22.1上的GLA基因突变,导致其编码产物α-半乳糖苷酶A(α-Gal A)出现错误折叠和修饰而使其活性降低或完全缺乏。当酶活性低于30%时,其作用底物三己糖酰基鞘脂醇(GL-3)及其衍生物脱乙酰基GL-3(Lyso-GL-3)的正常代谢受阻,进而在人体多种器官(如神经系统、肾脏、心脏、皮肤、肺、眼和耳等)中进行性贮积,最终引起一系列临床症候群[1]。临床分为经典型和迟发型。男女均可发病,男性多于儿童或青少年期起病,累及多个器官,酶活性显著下降;女性患者发病相对晚,可仅表现为单个器官损伤,甚至无症状。现对法布雷病的诊断及治疗进展进行阐述,旨在提高对法布雷病的诊疗及管理水平,从而改善预后。
法布雷病是一种罕见病,已被列入中国2018年5月颁布的首批121种罕见病目录。法布雷病在包括白人、亚洲人、西班牙人、黑人等几乎所有人种、民族中均有报道[1,2]。男性新生儿中法布雷病的发病率为1/117 000~1/40 000,随着新生儿基因筛查技术在临床上的应用和普及,其患病率可能比过去预期的要高得多,据报道,中国台湾地区男性新生儿的发病率为1/1 250,美国伊利诺伊州和华盛顿州新生儿分别为1/3 000和1/10 000[2,3]。
因致病基因位于X染色体上,所以一般情况下男性患者(半合子)较女性患者(杂合子)症状出现得更早、病情更严重,但并不是所有患者或所有症状均如此。
经典型法布雷病患者中,2/3的男性和1/3的女性患有血管角质瘤,是最早出现、最具特异性的临床特征,表现为单个或多组小而凸起的暗红色斑点,多分布于脐、手、膝盖、肘部和躯干,在青春期可延伸至生殖器区域。它们的数量和大小可随年龄的增长而增加,并与其他系统性病变的严重程度相关。皮肤受累的另一突出表现为出汗功能受损。多数患儿表现为少汗或者无汗,是由于鞘糖脂的沉积堵塞汗腺及影响交感运动神经纤维功能导致,表现为不明原因的高热和运动耐受性差,但也有少数患者存在多汗症[1]。
眼部受累是早期常见的症状,主要表现为角膜涡状混浊、血管迂曲、干眼症、后囊性白内障或症状性结膜淋巴管扩张。其中角膜涡状混浊具有诊断意义,临床可无症状,裂隙灯检查易诊断。眼部病变在经典型和迟发型法布雷病患者分别占70%[1]和30%[4]。
GL-3的沉积激活钙依赖性电压门,无髓鞘和髓鞘薄的小神经纤维易早期受累,表现为小纤维神经病变(small fiber neuropathy,SFN)。患者出现急或慢性疼痛、感觉障碍、乏力、出汗障碍、胃肠道功能紊乱、听力损害和焦虑等症状。其中,神经病理性疼痛最常见,男、女性患者的发病率分别为76%和64%,平均发病年龄分别为9.4岁和16.9岁[5]。
脑血管受累主要表现为卒中和短暂性脑缺血发作(transient ischaemic attacks,TIA)。卒中的发病率在女性约为4.3%,男性约为6.9%,多为缺血性卒中(87%)[6]。约2/3的患者在40岁左右出现继发于进行性微血管受累的脑白质高信号。也有患者表现为基底动脉扩张、认知障碍。
蛋白尿、肾小球滤过率下降最常见。镜下血尿、肾病综合征或肾小管酸中毒相对少见。约30%的患者在30岁左右进展至终末期肾病,肾脏受累与死亡率显著相关[2]。
40%~60%的法布雷病患者出现心脏受累,常见典型的左心室肥厚(LVH),且患病率随着年龄的增长而增加。在病因不明的肥厚型心肌病患者中,法布雷病的检出率高达0.9%~3.9%[1,7]。其他病变包括心律失常(室上性和室性心律失常、心房颤动、心动过缓),其中窦性心动过缓是最常见的心律异常;心肌病和心肌纤维化可导致传导异常和猝死;瓣膜疾病;微血管功能障碍(心肌缺血、心肌梗死)。心脏受累在经典型和迟发型法布雷病患者中相似。超声心动图和磁共振有助于诊断。
法布雷病患者可出现进行性或突发性听力受损(35.1%),约1/2的患者可出现眩晕,耳鸣也很常见[8]。
50%~60%的患者可出现胃肠道症状,腹痛、腹胀、腹泻、便秘、恶心、呕吐或假性梗阻综合征,常继发于肠道自主神经功能和血管病变。内窥镜检查一般正常,故以胃肠道症状为主的患者易延迟诊断。
法布雷病患者可过早出现骨质疏松症。呼吸系统病变有慢性支气管炎、呼吸困难、支气管哮喘等阻塞性肺功能障碍以及睡眠呼吸障碍等。
不同年龄段,临床表现可不一。2021年中国法布雷病诊疗专家共识进行了归纳总结,患者多在青少年时期出现症状,肾脏、心脏、脑是后期主要受累脏器[2]。
法布雷病是一种X连锁伴性遗传性疾病,需详细询问病史。家族中如有脑卒中或TIA、猝死、过早死亡、心肌病(尤其是肥厚型心肌病)、肾脏受累或透析、肢痛或不明原因的胃肠道症状,更需高度警惕法布雷病。
α-Gal A酶活性的测定是实验室检查的第一步。血白细胞(金标准)、血浆或干血纸片(DBS)均可用作α-Gal A酶活性的测定。干血纸片运输成本低,更实用,可作为临床筛查首选。
α-Gal A酶活性<3%,症状一般典型而且严重;酶活性介于3%~ 30%的患者,临床表现相对轻或发病较迟。但女性患者存在X染色体随机失活,α-Gal A酶活性水平不一,60%以上的女性患者α-Gal A酶活性在参考值范围内[2],需进一步行GL-3和Lyso-GL-3的测定及基因检测帮助诊断。
可通过测定因代谢障碍而产生的GL-3来帮助诊断。男性患者血浆和尿液中的GL-3水平显著高于健康人群,女性患者多处于正常范围。血浆和尿液中Lyso-GL-3的检测更为敏感和特异[9]。可通过DBS样本进行Lyso-GL-3检测。GL-3或Lyso-GL-3水平通常与临床表现严重性相关,经典型患者血、尿GL-3和Lyso-GL-3水平显著增高,迟发型患者GL-3和Lyso-GL-3水平则较低。男性患者血、尿Lyso-GL-3升高,女性患者在儿童期血浆Lyso-GL-3水平多正常,但随着年龄增长,40%~60%出现Lyso-GL-3升高[10]。成年女性α-Gal A酶活性和血浆Lyso-GL-3的测定均有助于疑似病例的诊断。
可进行肾脏、心脏、皮肤或神经组织活检,具有辅助诊断意义。光镜下可见受累组织细胞空泡化,细胞内见GL-3沉积。电镜下可见受累组织细胞胞质内充满嗜锇性"髓样小体",呈圆形或卵圆形,小体内部呈层状,类似洋葱皮或髓鞘结构,是溶酶体糖脂聚集的典型病理特征[2]。存在心脏病变的患者,必要时可行心内膜活检。但病理检查需有创操作,其必要性随着血尿生化及基因检测的发展有所下降。
位于Xq22.1的致病基因GLA突变的检测对明确诊断及筛查发现新患者有重要意义。根据家族史情况,推荐进行家系基因检测、绘制遗传图谱。目前已报道近1 000种GLA突变,但仅有80%左右患者可检测到致病基因突变或良性基因突变,需多方面综合分析。相同的基因突变可能存在不同表型。临床表型受年龄、性别及基因突变等多个因素影响。女性患者因存在X染色体随机失活,GL-3的沉积程度不一,故临床表现轻重不同。基因检测还有助于指导疾病治疗。现已研发合成的药物伴侣米加司他(Migalastat),对40%的基因突变患者有效[11]。
此外,酶活性低的法布雷患者,基因检测未发现外显子突变,应进行内含子突变检测、评估。
疑似法布雷病患者的诊断流程见图1。法布雷病临床表现缺乏特异性,需结合酶活性、生物标志物GL-3及Lyso-GL-3的测定、病理及基因检测等,尽可能避免误诊、漏诊。
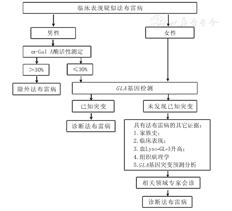
注:α-Gal A:α-半乳糖酶A;Lyso-GL-3:脱乙酰基三己糖酰基鞘脂醇 α-Gal A:α-galactosidase A;Lyso-GL-3:deacetylated globotriaosylcera-mides
法布雷病临床表现多样化,故需要与其他疾病相鉴别。在儿科群体中,需着重与其他系统性疾病鉴别,如系统性红斑狼疮、结缔组织病、皮肌炎、雷诺综合征、过敏性紫癜等[12]。
目前针对法布雷病特定的治疗方法有酶替代疗法(ERT)和药物伴侣。辅助治疗、心理支持和社会支持也必不可少。推荐多学科联合诊疗,制定合适的个体化治疗方案和管理策略,目标在于延缓疾病进展,改善生活质量,延长患者生命。
ERT自2001年起应用于临床,目前仍是法布雷病的主要治疗方法。现有的ERT药物有2种,均为静脉给药:阿加糖酶β(Fabrazyme,应用于年龄≥8岁,推荐治疗剂量1.0 mg/kg,每2周静脉输注1次)和阿加糖酶α(Replagal,应用于年龄≥7岁,推荐治疗剂量0.2 mg/kg,每2周静脉输注1次),仅阿加糖酶β被美国食品药品监督管理局(FDA)批准用于法布雷病的治疗。
目前主张尽早开始ERT,避免组织、器官不可逆损伤。2021年我国法布雷病诊疗专家共识也分别列出儿童和成人ERT起始治疗指征[2]。经典型男性患者,一旦确诊,即可开始ERT。对于经典型女性患者及迟发型男性患者,出现法布雷病特异性症状,或有肾脏、心脏、神经系统等受累的实验室或影像学证据,应尽早开始ERT。其中经典型患者的诊断是基于GLA基因突变,α-Gal A残余酶活性显著下降甚至缺乏,以及至少存在以下一种情况:血管角质瘤、角膜涡状混浊或高水平的Lyso-GL-3。ERT存在一些缺点:(1)靶器官严重损伤时ERT效果甚微;(2)ERT常触发抗α-Gal A抗体的产生,尤其见于经典型男性患者,研究表明IgG抗体在开始ERT的3~6个月内产生,导致疗效欠佳[13];(3)ERT的年花费巨大。
针对抗α-Gal A抗体的产生,研究报道应用环磷酰胺与静脉注射免疫球蛋白或利妥昔单抗联合治疗,有助于降低抗药物抗体(anti-drug antibodies,ADAs)滴度[14]。
药物伴侣于2016年在欧洲被引入,是一种通过口服用药治疗法布雷病的可行性方案。目前批准上市的药物是米加司他(1-脱氧半乳糖胞苷)。米加司他可纠正酶的错误折叠,稳定功能失调的α-Gal A,并促进其转运至细胞溶酶体内。一旦进入溶酶体,适当的环境(pH值较低、具有较高浓度的鞘糖脂底物)下,米加司他便与α-Gal A解离,从而恢复酶活性,清除法布雷病特征性鞘糖脂底物的积聚。
米加司他适用于部分错义突变的成人患者和≥16岁青少年患者。推荐的给药方案为:123 mg隔日1粒,空腹口服(服药前、后2 h内禁食)。不推荐应用于估算的肾小球滤过率(eGFR)<30 mL/(min·1.73 m2)的患者。目前至少有359个错义突变可用米加司他治疗,该药有望成为独立口服治疗法布雷病的一线疗法[15]。
Hughes等[16]建议下列情况可选用米加司他,而非ERT:≥16岁,部分错义突变,eGFR>30 mL/(min·1.73 m2),计划不孕的女性,隔日口服药物依从性好,或者对ERT不敏感的患者。研究表明米加司他在改善左心功能方面优于ERT。此外,米加司他可以透过血脑屏障,在治疗或改善法布雷病患者神经系统病变方面亦有很好的应用前景。
另一种口服治疗药物Ibiglustat(GZ/SAR402671或Genz-682452,Venglustat),是一种新型的葡糖基分支合酶的变构特异性抑制剂,能够跨血脑屏障,主要用于神经系统病变的治疗。二期临床药物试验显示[17],与单药治疗相比,与ERT联合使用时,Ibiglustat能进一步减少底物的积累;建议早期用药,以防止不可逆的病理损伤。
在引入ERT之前,法布雷病的治疗局限于对症治疗,包括止痛、降尿蛋白、抗心律失常等。目前抗血小板聚集、血管紧张素转换酶抑制剂、β受体阻滞剂、他汀类药物等仍用于辅助治疗,以预防心脑血管危险因素的发生。
所有作者均声明不存在利益冲突

























