
面对各种因素导致的DNA损伤,细胞有一套应答修复机制。其中细胞周期阻滞在DNA损伤应答修复中扮演了重要角色,为修复受损DNA创造了条件。关于细胞周期调控的研究集中于细胞周期蛋白依赖性蛋白激酶(CDK)与细胞周期检查点等方面。在DNA损伤修复过程中,损伤位点募集的磷脂酰肌醇-3-激酶样激酶(PIKKs)可引起细胞周期检查点相关蛋白的激活,导致细胞周期阻滞。在碱基切除修复、核苷酸切除修复、错配修复、DNA双链断裂修复等常见的DNA损伤修复途径中,招募的损伤修复相关蛋白在细胞周期调控中也起到一定的作用。因此综述了主要DNA损伤修复形式与细胞周期阻滞之间的关系及研究进展。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
DNA是遗传物质的载体,决定了不同生物体的生物性状。细胞面临的多种内源性和外源性因素可引起不同形式的DNA损伤,内源性因素包括氧化损伤、DNA复制错误、DNA甲基化修饰等[1];外源性因素包括电离辐射、紫外线辐射等[2]。DNA损伤可引发细胞产生一系列反应:细胞感知DNA损伤并激活细胞周期检查点导致细胞周期阻滞,招募相关DNA损伤修复蛋白至损伤位点,激活多条细胞存活/死亡途径来拯救受损细胞或清除严重失调的细胞。
20世纪20年代,Wilson提出细胞周期的概念,认为细胞周期处于所有生物体生长、发育和遗传的核心位置[3]。20世纪50年代,关于细胞周期的研究成为热点。细胞周期是指分裂的细胞从一次分裂完成到下一次分裂结束所经历的过程,分为间期和分裂期(M期)[4]。间期又分为DNA合成前期(G1期)、DNA合成期(S期)、DNA合成后期(G2期)。DNA损伤后可在G1或G2期引起细胞周期阻滞,或延缓S期复制,以促进DNA损伤修复。本文旨在阐述DNA损伤修复与细胞周期调控相关的研究,为细胞周期阻滞与DNA损伤修复相互作用关系提供研究思路。
细胞周期蛋白依赖性蛋白激酶(cyclin-dependent kinase, CDK)是控制细胞周期的核心物质。细胞周期蛋白(cyclin)是CDK的正调控因子,细胞周期蛋白依赖性蛋白激酶抑制剂(cyclin-dependent kinase inhibitor, CKI)是CDK的负调控因子[5]。cyclin作为蛋白激酶复合体的调节亚基在细胞周期中进行更替,与相应的CDK结合,调节细胞周期进程。在G1期,CDK4和CDK6分别与cyclin D1、cyclin D2、cyclin D3相互作用[6];在G1/S期,CDK2与cyclin E相互作用[7];在S期,CDK2与cyclin A相互作用[8]。cyclin B的表达开始于S期,在G2/M期达到高峰,M期结束后被降解[9];在G2/M期和M期,CDK1分别与cyclin A和cyclin B相互作用[6]。
正常条件下细胞是按规律生长的,细胞发生周期阻滞或继续增殖分裂取决于各种复杂信号如何对细胞周期的核心机制进行调控,其中视网膜母细胞瘤抑癌蛋白(Rb)与转录因子E2F组成的双稳态机制在G1/S期发挥了重要作用(图1)。E2F可介导DNA复制以及细胞周期相关蛋白的转录,当细胞处于静止状态时E2F与Rb结合并被抑制。在正常的细胞周期循环过程中,CDK4/6与cyclin D(D1、D2、D3)结合后可磷酸化Rb,抑制Rb功能并启动E2F转录。另外E2F转录激活后又促进了CDK2与cyclin E(E1、E2)的结合,该复合物进一步导致Rb磷酸化,起到双稳态作用[10]。
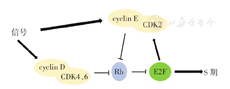
Rb—视网膜母细胞瘤抑癌蛋白;cyclin—细胞周期蛋白;CDK—细胞周期蛋白依赖性蛋白激酶;S期—DNA合成期
为了避免将改变的基因组传递给子细胞,细胞已进化出了复杂的检查点机制来阻止细胞周期进程从而促进DNA修复,或在损伤不可修复的情况下促使细胞死亡。细胞周期检查点主要分为细胞周期G1、S、G2检查点以及有丝分裂纺锤体组装检查点(spindle assembly checkpoint, SAC)[11,12]。G1期检查点主要由共济失调毛细血管扩张突变蛋白(ataxia-telangiectasia mutated protein, ATM)-细胞周期检查点激酶2(cell cycle checkpoint kinase 2,Chk2)通路介导,能引起持续甚至永久的细胞周期阻滞[13];S期检查点由ATM/ATM与Rad3相关蛋白(ATM and Rad3-related protein,ATR)控制,一方面经由Cdc25A介导的级联降解起作用,另一方面经由ATM介导Nbs1的多个位点的磷酸化起作用[14];G2期检查点可阻止有丝分裂的启动,修复G2期以及之前未被修复的DNA损伤,主要由ATR-Chk1通路介导;由BubR1、Bub3和Mad2相关蛋白与APC/C共激活子Cdc20组成的有丝分裂检查点复合体(MCC)可启动SAC[15]。SAC的存在确保了姐妹染色单体正确连接后才发生染色体分离。
细胞分裂期中未被修复的DNA损伤可导致非整倍体复制和突变的发生。DNA损伤导致的细胞周期阻滞最终表现为CDK的失活,当DNA损伤修复完成后,CDK被重新激活从而恢复细胞周期循环[16]。此外在人类癌症细胞中检查点机制的失活很常见,被认为是肿瘤形成的标志之一[14],且电离辐射或某些治疗癌症的化学治疗(化疗)药物等也会激活细胞周期检查点。
由感受器、中介因子、转导因子和效应器等构成的DNA损伤应答反应可起到激活DNA损伤修复、诱导细胞凋亡、导致细胞周期阻滞等作用[17]。在DNA损伤应答反应过程中,磷脂酰肌醇-3-激酶样激酶(phosphatidylinositol-3-kinase-like kinases, PIKKs)家族蛋白起主导作用,其可启动一系列磷酸化级联反应,减缓或暂停细胞周期进程,以促进DNA的损伤修复[18]。
肿瘤抑制基因p53在启动细胞周期阻滞方面具有重要作用,Chk1和Chk2也可通过不依赖于p53的途径来启动细胞周期阻滞[19]。另外,细胞在某个时期停留过久会产生不良后果,如上皮细胞在G1/G2期阻滞过久会导致衰老和纤维化表型的形成[20],因此损伤因素消失后迅速逆转该过程十分重要。
G1期阻滞可由ATM-Chk2途径调控。ATM是一种苏氨酸/丝氨酸蛋白激酶,属于PIKKs家族成员,该家族成员还包括ATR和DNA依赖性蛋白激酶催化亚基(the catalytic subunit of DNA-dependent kinase, DNA-PKcs)[21]。ATM可磷酸化Chk2的Thr68位点,激活的Chk2可磷酸化Cdc25A(Cdc25B和Cdc25C也存在部分激活),产生与14-3-3蛋白结合的位点,被转运出细胞核。蛋白酶体降解核内转运出来的Cdc25A,造成CDK2失活,从而使细胞周期阻滞在G1期[22]。另外DNA损伤发生后,损伤部位可募集Mre11/Rad50/Nbs1(MRN)复合物,磷酸化ATM的Ser1981位点,并通过肿瘤抑制因子p53结合蛋白1(p53 binding protein 1, 53BP1)激活磷酸化组蛋白H2AX[23]。在电离辐射作用下,ATM还可磷酸化激活转录因子2(ATF2)的Ser490和Ser498位点,导致ATM、MRN复合物和磷酸化组蛋白H2AX的快速聚集[24]。
从G1期结束到S期、G2期,对细胞周期的控制由ATM-Chk2途径转换为ATR-Chk1途径。DNA损伤修复反应发生时,被招募来的ATM可进一步招募SMC1等蛋白分子,SMC1是S期的检查点蛋白,其Ser957和Ser966位点会被ATM磷酸化从而发挥细胞周期调控功能[25]。CDK2依赖性磷酸化可促进ATR和Chk1的活化,将ATR和Chk1的激活限制在S期和G2期[26]。另外增殖细胞核抗原(PCNA)相关的CRL4-Cdt2泛素连接酶抑制了p21的积累,随着p21的不断降解,Wee1激酶在S期表达,通过磷酸化CDK2来抑制其活性,使细胞周期阻滞在S期[27]。
G2/M检查点由ATR/Chk1触发,防止细胞携带损伤的DNA进入M期。CDK1是M期的主要激酶,正常情况下,Cdc25C可使CDK1的Thr14和Tyr15位点去磷酸化,活化CDK1-cyclinB,使细胞进入M期[28]。DNA受到损伤时,招募的ATR可在中介蛋白Claspin的作用下磷酸化Chk1的Ser317和Ser345两个位点[29]。活化的Chk1磷酸化Cdc25C的Ser216位点,使其与14-3-3蛋白结合,使细胞周期阻滞在G2期。Chk1的稳定性与其自磷酸化状态有关,在细胞增殖过程中Chk1通过基础活性来维持检查点的功能。此外Wee1激酶和Myt1激酶可磷酸化CDK1的Thr14和Tyr15位点,在S期和G2期抑制CDK1的活性,使细胞不能进入M期[30]。在DNA急性损伤和应激反应消退后,泛素-蛋白酶体降解系统启动导致Chk1及Claspin的降解,终止了检查点途径[31,32]。
在癌细胞中G1/S检查点常存在缺陷,如p53和Rb突变所致缺陷,此时癌细胞可依赖S期和G2/M期检查点来修复之前遗留的DNA损伤,而S期检查点可能仅有助于减缓而非停止细胞周期。由于携带DNA损伤的癌细胞一般可通过S期检查点,停止于G2/M检查点[33],因此G2/M期检查点的激活是癌细胞完成DNA损伤修复的重要保障。
DNA损伤修复途径包括碱基切除修复(base excision repair,BER)、核苷酸切除修复(nucleotide excision repair, NER)、DNA错配修复(mismatch repair,MMR)以及DNA双链断裂(double-strand break, DSB)修复等[34]。DNA损伤修复过程与细胞周期阻滞同时发生,这些修复过程是维持细胞遗传稳定性的关键,因此DNA损伤修复信号通路中的蛋白分子在细胞周期进程的调控中通常也发挥作用。
BER途径可对活性氧、电离辐射或紫外线辐射等产生的碱基损伤进行修复,主要发生在G1期[35]。DNA糖基化酶可识别并切除受损碱基,其中单功能糖基化酶产生的无碱基位点参与了短片段修复途径,而双功能糖基化酶则介导了长片段修复途径[35]。此外,聚腺苷二磷酸核糖聚合酶-1 [poly(ADP-ribose)polymerase-1, PARP-1]被证明是通过BER途径修复单链断裂和受损碱基所必需的[36]。PARP-1含有3个主要结构域:锌指结构的基序组成氨基末端DNA结合结构域、包含BRCT结构域的中央自修饰结构域和高度保守的羧基末端催化结构域[37],它们共同介导PARP-1对不同DNA损伤类型的反应。
NER是重要且复杂的DNA损伤修复系统,可修复紫外线、化学试剂或活性氧引起的大体积DNA损伤和DNA链交联。NER缺陷可引起着色性干皮病和科克因综合征等遗传性疾病[38]。NER分为修复速度较慢的全基因组修复途径和修复速度较快的转录偶连修复途径。后者负责修复转录活跃的DNA链的损伤,而前者负责修复基因组其余部分的损伤,包括非转录链的损伤及沉默染色质区域的损伤。关于NER与细胞周期检查点的报道较少,但近来有研究结果显示,同时靶向NER和细胞周期检查点的肽纳米复合物可在肺腺癌中表现出更好的化疗效果[39]。
MMR途径由蛋白MSH2~MSH6识别并结合错配碱基来开启,是细胞应对DNA复制错误并维持基因组稳定性的途径之一。MMR功能的丧失会增加基因突变频率,与肿瘤发生相关[40]。在G2期,MMR可通过激活Chk1的Ser317位点导致细胞周期阻滞[41],并可响应6-硫鸟嘌呤或N-甲基-N'-硝基-N-亚硝基胍诱导的DNA损伤[42]。当使用ATR激酶抑制剂抑制G2期检查点时,可减轻N-甲基-N'-硝基-N-亚硝基胍诱导的Chk1激活和天冬氨酸蛋白酶的积累,同时增加DSB损伤和细胞死亡。
细胞暴露于电离辐射和某些化学物质后会发生DSB[43]。在哺乳动物细胞中DSB修复的两条主要途径是同源重组(homologous recombination, HR)和非同源末端连接(non-homologous end joining, NHEJ)。NHEJ发生在细胞周期的各个阶段,而HR主要发生在S/G2期,以姐妹染色单体作为DSB修复模板[44]。
HR修复通路主要发生在S期,其中MRN复合物起到了重要作用。MRN复合物结合至DSB部位募集ATM等信号分子,催化对断裂链5'末端的处理和招募长链核酸酶Exo1和Dna2[45,46]。在DNA核酸内切酶CtIP的帮助下,通过MRN的内切酶活性切除冗余末端,然后Exo1或Dna2延伸切除区域并产生3'单链DNA[47]。MRN复合物中Nbs1的FHA结构域和BRCT重复结构域中存在双重磷酸化肽结合区域,在细胞周期检查点和DNA修复信号转导中均起着关键作用[48]。
在NHEJ修复通路中,53BP1发挥向受损染色质募集分子元件的作用[49]。Ku是第一个被募集的二聚体复合物(包括Ku70和Ku80),其作为连接点再招募DNA-PKcs等其他分子[50]。有研究结果发现,53BP1可通过与ATM协同作用,调控Eca-109细胞中p53、Chk1和Chk2的表达导致细胞周期阻滞,在DNA损伤检查点响应中起着重要作用[51,52]。在有丝分裂出现长时间明显缺陷的情况下,53BP1和去泛素化酶USP28作为p53上游的重要调控分子,可引发p21依赖的细胞周期阻滞,而53BP1和USP28的功能与阻止有丝分裂的干扰或延迟有关,可提高有丝分裂的效率[53]。在S期,cyclin-CDK对Ku70的周期性磷酸化阻止了Ku与复制起点的相互作用,而有丝分裂结束时的去磷酸化则促进了Ku在G1期中修复DSB的功能[54]。DNA-PKcs的激酶活性在NHEJ中十分重要,已报道了DNA-PKcs的多种底物[55]。在ATR缺乏的情况下,DNA-PKcs可促进Chk1的激活[56],提示DNA-PKcs与下游检查点激酶之间可能存在复杂的联系,但DNA-PKcs在细胞周期调控中的其他磷酸化底物蛋白尚不清楚,值得进一步的探索。
随着对细胞周期研究的深入,人们发现细胞周期调控在DNA损伤修复中起到了非常重要的作用。各种CDK与细胞周期检查点机制共同精确地调控细胞周期进程,G1/S/G2期的细胞周期阻滞为DNA损伤修复赢得了时间。DNA损伤修复过程中的DNA损伤修复蛋白与细胞周期检查点机制的相互作用还有待深入研究:如DNA损伤修复反应导致细胞周期阻滞是否存在阈值效应,以及其是否依赖于细胞类型或表观遗传条件等。此外关键时期细胞周期阻滞的激活和失活也是癌症干预治疗的目标之一,关于Chk抑制剂的治疗窗口以及预测治疗效果的标志物还需探究。研究细胞周期阻滞相关通路在DNA损伤修复中的作用对于深入研究电离辐射损伤修复、放化疗机制、肿瘤治疗等也具有指导意义。
所有作者均声明不存在利益冲突

























