
尽管雄激素剥夺疗法对于进展性和转移性前列腺癌的治疗有一定效果,但患者不可避免产生耐药,发展为去势抵抗性前列腺癌(CRPC)。其中,雄激素受体(AR)信号通路再激活是CRPC进展的关键驱动因素。近年来,蛋白水解靶向嵌合体(PROTACs)通过与靶蛋白、E3泛素化连接酶形成三元复合物对靶蛋白高效特异性降解,在CRPC领域得到迅速发展,其中ARV-110,ARV-471、AR-LDD均已进入临床试验阶段。然而,PROTACs分子在研究进程中面临诸多困难。随着纳米医学的发展,其与PROTACs联合应用或将对降低CRPC患者AR的表达,解决病情进展起到重要作用。本文就AR与CRPC的治疗,以及PROTACs的作用机制及特点,PROTACs在去势抵抗性前列腺癌研究领域的应用情况及现存问题等进行了综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
前列腺癌是目前全球男性第二常见的恶性肿瘤,也是我国近年来发病率增长最快的恶性肿瘤之一[1]。自1941年Huggins 和Hodogs发现前列腺癌存在雄激素依赖性,降低雄激素水平和(或)阻断雄激素受体(androgen receptor,AR)可抑制前列腺癌细胞生长,即雄激素剥夺疗法(androgen deprivation therapy,ADT),是目前治疗进展性和转移性前列腺癌指南推荐的标准方式[2, 3]。但患者面临高死亡率,目前尚无治愈的方法。故本文将蛋白水解靶向嵌合体(proteolysis-targeting chimeras,PROTACs)与纳米技术在去势抵抗性前列腺癌(castration resistant prostate cancer,CRPC)治疗中的发展现状及展望作如下综述。
ADT根据作用机制和靶点的不同,分为去势治疗和抗雄治疗两大类。抗雄治疗即药物与雄激素竞争结合AR,阻止靶器官摄取雄激素,从而抑制肿瘤生长;去势治疗即祛除产生睾酮器官或抑制产生睾酮器官的功能,抑制睾丸分泌雄激素,包括手术去势和药物去势[3]。CRPC是经过初次持续ADT产生耐药性后进展的前列腺癌,其中位进展期为18~24个月[4]。
AR信号通路再激活是CRPC进展的关键驱动因素[5]。单纯ADT治疗仅能阻断睾丸来源的雄激素,对于肾上腺和前列腺肿瘤自身合成的雄激素不能阻断。尽管血清睾酮处于去势水平(<1.7 nmol/L),但肿瘤自身合成雄激素能力提高。此外,AR基因通过突变、过表达和剪切变异等可再激活AR信号通路[6]。因此对于CRPC,如何有效降低AR的表达,加强对AR药物耐药性的探索,是解决CRPC阶段患者病情进展的关键问题。
在癌症治疗方面,降解蛋白质比抑制蛋白质合成具有更大疗效[7]。靶向蛋白质降解(targeted protein degradation,TPD),即消除靶蛋白(protein of interest,POI)的过程,在癌症治疗领域具有可观的发展前景[8]。PROTACs的概念最早在2001年由Crews和Deshaies提出,并成功利用该技术对甲硫氨酰氨肽酶2(MetAp-2)进行降解[9]。PROTACs是一种具有异源双功能的小分子降解剂,由3个元素构成:两端分别为POI结合配体和E3泛素连接酶(ubiquitin-ligase enzymes E3)配体,中间为连接头(linker)。当PROTACs进入细胞后,通过柔性化学接头识别并结合POI,特异性募集E3泛素连接酶,形成稳定的“POI-PROTACs-E3泛素化连接酶”三元复合物。E2泛素结合酶(ubiquitin-conjugating enzyme E2-UCE)与E3泛素连接酶共同作用将POI聚泛素化标记后,通过细胞内现存的泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)特异并高效地使POI降解,从而治疗疾病[10](图1)。
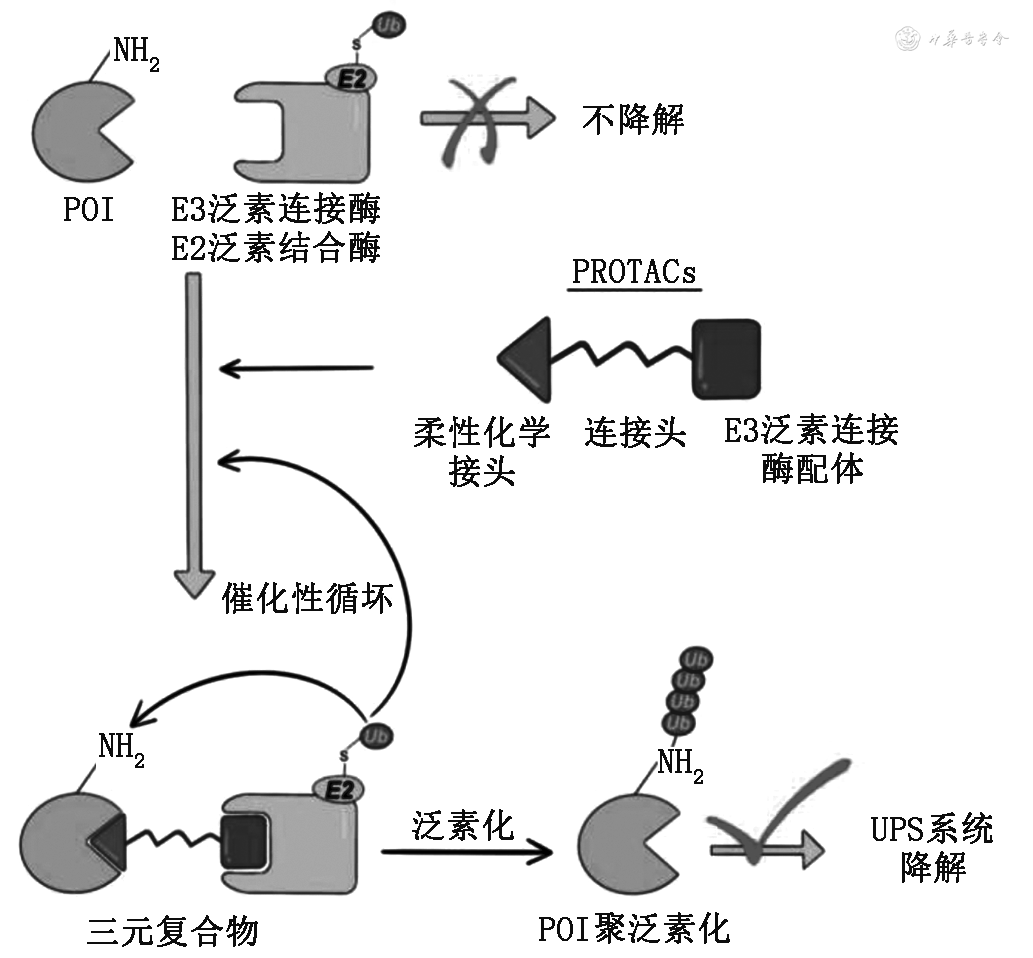
与传统的小分子蛋白抑制剂相比,PROTACs有诸多优点。首先,基于开放性的分子设计,PROTACs在开发时可选择不同种类的POI配体及E3泛素连接酶配体,不仅靶向降解蛋白质种类丰富,如激酶类、溴结构域和末端外结构域(bromodomain and extraterminal domain,BET)、蛋白类、核受体等[11],其对靶蛋白的选择性也进一步提高。目前文献报道PROTACs募集的E3泛素连接酶配体主要有羟脑苷脂(cereblon,CRBN)、希佩尔林道肿瘤抑制基因(von Hippel-Lindau、VHL)、细胞凋亡抑制蛋白(cellular inhibitors of apoptosis proteins,cIAP)和鼠双微体2基因(murine double minute2,MDM2)4种类型,其中使用率高、效果好的主要是CRBN和VHL两种,可通过类似药物小分子的E3泛素连接酶进行招募[12]。CRBN的配体为免疫调节酰亚胺(immunomodulatory imide,IMiDs)如来那度胺及类似物,VHL配体主要是VHL-L。其次,不同于传统小分子“占用驱动”作用模式,PROTACs作用特点是“事件驱动”型,即在泛素化标记一个POI后,与POI和E3泛素连接酶解离,寻找下一个POI进行标记,如此形成催化性循环(图1),利用少量的PROTACs分子便可实现对大量POI进行降解目的。更重要的是,“POI-PROTACs-E3泛素化连接酶”三元复合物的形成是高效降解靶蛋白的关键,弱化了PROTACs分子与POI或E3泛素连接酶亲和力和结合牢固程度的影响[13]。基于以上优点,在针对以往不可成药的靶点中,PROTACs应用前景可观。
有研究报告[14]显示,到2021年底,至少将有10种PROTACs进入临床试验,其中包括以AR蛋白作为靶蛋白,针对去势抵抗性前列腺癌设计的ARV-110(Androgen receptor degrader临床Ⅱ期),ARV-766(Androgen receptor degrader)、AR-LDD(Androgen receptor degrader临床Ⅰ期),有望为CRPC治疗提供新途径。
Neklesa 等[15]报道了基于CRBN配体合成的口服蛋白质降解剂ARV-110,选择性靶向结合并降解AR蛋白,阻止AR靶基因的表达和信号传导。前期研究发现,ARV-110对AR降解率高达95%,同时抑制AR过表达肿瘤细胞的增殖。在恩杂鲁胺耐药模型中ARV-110能明显抑制肿瘤生长;而在恩杂鲁胺敏感的模型中,ARV-110可导致前列腺特异性抗原(prostate specific antigen,PSA)降低。在一期临床试验(NCT03888612)纳入的15例AR野生型CRPC患者中,2例(13%)患者PSA降低50%。在另外5例带有T878A或H875Y突变亚群的患者则表现出更强的抗肿瘤活性,4例患者PSA水平降低30%以上,2例患者PSA水平降低50%以上,其中1例经实体瘤疗效评价标准(Response Evaluation Criteria in Solid Tumors,RECIST)证实为部分缓解(partial response,PR)肿瘤体积缩小至20%。目前,ARV-110的Ⅱ期临床实验正在进行。此外,相关文献报道了基于VHL配体合成的PROTAC-3[16],ARD-69[17],ARD-61[18]、ARV-771[19];基于cIAP配体合成的特异性和非遗传性IAP-依赖性蛋白质擦除器(specific and nongenetic IAP-dependent protein erasers,SNIPER)如ARCC-4等[20]。尽管这些复合物在前期体外模型中均表现出对AR不同程度的降解,但PROTAC仍有许多技术问题亟待解决。
首先,“POI-PROTACs-E3泛素化连接酶”作为核心生物学事件,其稳定性对于PROTACs诱导降解效率至关重要。研究表明,PROTACs分子浓度分布符合正态曲线,浓度过高或过低时,三元复合物生成均受到影响[21]。这也符合由于抗原抗体比例不合适导致的钩状效应(hook effect)[22],即PROTACs浓度过高时竞争性形成POI-PROTACs、PROTACs-E3泛素连接酶、POI-E3泛素连接酶二元复合物,影响PROTACs的工作效率。其次,目前没有针对PROTACs可靠的生物活性评价模型。从作用机制来看,PROTACs相对复杂的三联体结构及催化特性使得其在组织中可能不遵循相位型(phase,PH)分布理论,有出现靶点介导的药物处置现象(target mediated drug disposition,TMDD)的可能性,即仅产生一种类型的药物靶标复合物理论,导致药代动力学(pharmacokinetics,PK)和药效动力学(pharmacodynamics,PD)复杂化[23]。用传统方法测定PROTACs的曲线不可取,因此在吸收、分布、代谢、排泄以及药物毒性和给药剂量方面都无法准确评估。此外,相关研究报道Linker的长度与两端配体的连接位点都会影响PROTACs的选择性和降解能力[24],可能是由于泛素化过程中不同的POI与E3泛素连接酶所需空间不同,PROTACs分子通过“自我折叠”,使两者产生蛋白质-蛋白质相互作用(protein-protein interactiojn,PPI),进而影响PROTACs分子细胞渗透率、组织分布等。目前对linker部分的长度和组成设计原则缺少指导方向。此外,脱靶毒性是对PROTACs安全性最大的担忧。据报道,CRBN配体上的邻苯二甲酰亚胺和戊二酰亚胺官能团不稳定,存在水解或变构的问题[25]。脱靶蛋白降解可能误伤其他正常组织,即所谓的旁观者效应(bystander effect,BE)。并且PROTACs脱靶效应在前期毒性筛选中不易检测追踪,因此增加了后期药物研发安全性风险[26]。
随着纳米技术和纳米医学的发展,一系列纳米材料出现在PROTACs治疗领域,例如纳米药物递送系统(nanoscale drug delivery system,NDDS)通过包封或吸附手段与小分子药物形成药物纳米颗粒来递送药物和治疗[27]。理想的纳米材料能将PROTACs分子包裹在颗粒中或吸附在其表面,通过靶向分子与细胞表面特异性受体相结合,并由细胞摄取进入胞内。实体瘤的高通透性和滞留效应(enhanced permeability and retention effect,EPR)是纳米载体在肿瘤治疗领域受到高度关注的重要原因,即肿瘤组织血管内皮间隙和窗孔较大,结构不完整且无功能性淋巴回流,纳米载体可选择性透过血管并积蓄于瘤体内,是纳米药物递送系统实现被动靶向性的纳米生物结构基础[28]。纳米药物递送系统具有高载药率、被动靶向功能、药物控制释放、生物利用率高而毒性低等诸多优点。对于PROTACs分子浓度控制、细胞渗透率及组织分布、脱靶毒性等问题,与纳米技术结合成为未来研究重点。
纳米金(gold nanoparticle,GNP)是直径在0.5~200 nm的金超微粒子,通过形成金-硫(Au-S)配位键,含巯基的药物可以在纳米金表面自行组装,形成稳定的纳米金药物递送系统。纳米金比表面积大,可负载大量药物,是高效的抗肿瘤载体[29]。Wang等[30]报道了一种基于纳米金的多头PROTACs新型复合物Cer/Pom-PEG@CNP,用于治疗间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)突变的非小细胞肺癌(non-small-cell lung cancer,NSCLC)。其中,色瑞替尼(Ceritinib)和泊马度胺(Pomalidomide)分别作为POI配体与E3泛素连接酶配体,CRBN作为E3泛素连接酶,并通过聚乙二醇(polyethylene glycol,PEG)2000形成金-硫配位键偶联到直径约30 nm的纳米金表面。体外试验中,透射电子显微镜(transmission electron microscopy,TEM)显示纳米药物递送系统与POI共同定位于细胞质中,并在给药24 h后保持稳定,以剂量和时间依赖的方式展示出强大的POI降解能力。结果显示,与单纯PROTACs复合物相比,纳米金药物递送系统实现对药物的可控释放,降低脱靶毒性。除PEG外,利用人血清白蛋白(bovine serum albumin,BSA)修饰于纳米金表面能提升其生物相容性,增加肿瘤细胞对纳米金的摄取[31];而球形结构的DNA或RNA寡核苷酸修饰能增强细胞膜介导的内吞作用[32],从而提升其透膜能力。
纳米材料除了作为载体靶向递送PROTACs分子外,其材料本身的特殊理化特征也可发挥协同作用,有效抑制肿瘤进展。光动力学疗法(photodynamic therapy,PDT)是指利用光能激发光敏剂(photosensitizer,PS)如近红外光(near infrared light,NIR)等到激发态,将能量传递给肿瘤周围的氧,生成活性氧(reactive oxygen species,ROS)如超氧化物(Superoxide)或单线态氧(1O2)等,进而产生毒性、损伤肿瘤周围血管和组织,造成肿瘤细胞缺血性死亡[33]。Zhang等[34]报道了一种用于可激活光免疫代谢癌症治疗的半导体聚合物纳米PROTACs(SPNpro)。吲哚胺 2,3-二氧合酶(IDO,Indoleamine 2,3-dioxygenase)抑制剂NLG919和组织蛋白酶B可切割片段(CatB-cleavable segment)分别作为POI配体与E3泛素连接酶配体,VHL作为E3泛素连接酶,通过PEG2000偶联在纳米载体上。体内实验证实,SPNpro可在NIR照射下产生单线态氧(1O2)以消除肿瘤细胞,并诱导肿瘤相关抗原释放;另一方面,仅在CatB特异性激活后SPNpro才可启动,IDO靶向降解阻止色氨酸(tryptophan,Trp)分解代谢为犬尿氨酸(kynurenine,Kyn)。二者发挥协同作用诱导树突状细胞(dendritic cells,DCs)成熟并促进T细胞活化,上调T细胞免疫反应,增强抗肿瘤作用。此方法可使药物更精准靶向治疗至瘤体,降低用药剂量,减少了超剂量PROTACs对正常组织、细胞的不良反应。
CRPC的治疗是前列腺癌领域研究的热点和难点,包括内分泌治疗、化疗、分子靶向治疗及免疫治疗等多种治疗手段。尽管如此,进展至此阶段的患者预后仍然较差。目前,进一步有效抑制AR的表达以及克服ADT药物耐药性的新方法亟待开发。PROTACs分子有靶向性强、降解效率高、可靶向“不可成药”靶点、克服耐药性等诸多优势。目前,基于PROTACs技术设计的新型复合物在CRPC领域有了多项研究,其中ARV-110、ARV-766、AR-LDD进入临床试验阶段,但其三元复合物形成困难、缺少可靠的生物活性评价模型、linker选择问题和脱靶毒性等缺点不容忽视。
近年来,纳米药物递送系统在生物医疗中发挥着重要作用,与PROTACs技术联合应用,为探索CRPC治疗新方法开辟道路。首先,纳米药物递送系统可靶向给药,包括肿瘤、炎症等利用脉管系统管径差到达疾病组织的被动靶向运输[35],以及通过特定配体-受体相互作用使药物与靶细胞相结合的主动靶向运输[36]等。在保证药物作用的前提下,减少PROTACs分子的脱靶毒性。其次,对于半衰期较短的药物,通过调控纳米材料表面性能使其PEG化、电中性等达到“隐身”效果而在肿瘤细胞附近脱去PEG、携带正电荷等方法获得黏附细胞能力,延长PROTACs药物作用时间[37]。另外,纳米药物递送系统可实现多样性和智能性。除了携带PROTACs分子外,还可同时携带多肽、核酸等生物活性物质等。不仅如此,纳米药物递送系统还可通过改变膜转运机制,增加药物对生物膜的透过性,更有利于药物的吸收与胞内发挥作用[38]。
然而,具有较长半衰期的持久性纳米颗粒在生物体内难以降解,根据国际标准制定机构的规定[39],其配方要求需要的最小测量值包括尺寸、zeta电位和溶解度,均需要严格临床评估。另外,纳米药物递送系统的体内性能包括生物相容性、生物安全性、药代动力学及药效动力学等,必须通过动物模型及药代动力学模型进行准确评估,但能准确反映人类恶性肿瘤的实验模型目前仍较为缺乏[40]。此外,纳米药物递送系统在大规模和重复性制备过程中可能伴随配方参数的优化、甚至工艺改变,但完备的临床评价和监测管理系统尚不完善[41]。虽然纳米药物递送系统的研发存在诸多挑战,但这都不能阻碍科研人员在相关领域不断研究和改进。随着纳米科技不断发展,相信医用纳米材料凭借其靶向性更强、不良反应更小且疗效更佳的优势将成为未来基础研究和临床应用的重点。同时,纳米技术联合抗肿瘤的治疗也会在精准医疗及转化医学的道路上不断前进。
所有作者均声明不存在利益冲突

























