
对1例以糖尿病起病的家族性部分脂肪营养不良综合征4型(FPLD4)患儿临床特征和基因变异特征进行回顾性分析。患儿,男,13岁5个月,糖尿病病程3年余,于2021年11月10日第4次入住苏州大学附属儿童医院,糖尿病病程中反复发生糖尿病酮症酸中毒逐渐出现脂代谢并发症,基因测序结果显示先证者及其母亲携带基因PLIN1 c.1325delG(p.G442Afs*99)和SPINK1 c.194+2T>C(p.?)双基因杂合变异。PLIN1为FPLD4的致病基因,c.1325delG变异目前尚未见文献报道。结合患儿临床表现、家族史及基因检测结果,诊断为FPLD4,同时携带基因SPINK1 c.194+2T>C(p.?)变异,可能使慢性胰腺炎发生的风险增加。本病例报道丰富了FPLD4的临床特点和基因型数据,基因检测方法的应用使患儿糖尿病分型得到了精准诊断,同时需警惕双基因突变对疾病进展的影响,对制定治疗方案和疾病管理具有重要意义。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
家族性部分脂肪营养不良综合征(FPLD)是脂肪营养不良综合征中的一种,于1974年被首次报道[1],发病罕见。目前已知的致病基因包括LMNA、PPAR-γ、ZMPSTE24、AKT2、PIK3R2、CIDEC、PLIN1、LIPE、ADRA2A和CAV1等。FPLD的脂肪分布特点为婴幼儿时期正常分布,随着年龄的增长,出现不同程度和部位的脂肪组织萎缩和糖脂代谢紊乱,后者包括高脂血症、糖尿病、胰岛素抵抗、多毛和多囊卵巢综合征等[2]。糖脂代谢紊乱的严重程度与脂肪萎缩的程度相关[3]。FPLD目前分为6种类型,PLIN1基因(OMIM 170290)为家族性部分脂肪营养不良综合征4型(FPLD4)的致病基因[2]。现回顾性分析1例PLIN1新发突变的儿童FPLD4病例,该患儿以糖尿病起病,反复发生糖尿病酮症酸中毒(DKA),并逐渐出现脂代谢异常。因其脂肪萎缩表现不典型,导致诊断不及时。本研究通过总结其临床特点和遗传学特征,以期提高临床医师对FPLD4的认识,加强临床管理,改善该类患儿的预后。
患儿,男,13岁5个月,糖尿病病程3年余。患儿10岁7个月和11岁5个月时分别因"多饮、多尿伴体重下降"于2018年12月19日及2019年10月16日至苏州大学附属儿童医院就诊(第1次及第2次就诊),诊断为"糖尿病,糖尿病酮症"。患儿体型轻度肥胖,颈部和腋下可见轻度黑棘皮,血清C肽正常,糖尿病自身抗体谱阴性,2次口服葡萄糖耐量试验及同步血浆胰岛素、C肽释放试验结果见表1。先后予二甲双胍和胰岛素治疗,平素未规律监测血糖,未定期随访及遵医嘱用药。13岁时因"腹痛2周,呕吐0.5 d"第3次入院(2021年5月17日)。近3年患儿未严格遵循糖尿病饮食,此次入院前有进食蛋糕病史。患儿系第1胎,第1产,足月顺产,出生无窒息抢救史,出生体重3 000 g,既往无其他慢性疾病史。父母非近亲结婚,母亲33岁(妊娠时)诊断糖尿病,体型匀称,糖尿病饮食,平素口服二甲双胍控制血糖,空腹血糖波动在7 mmol/L左右,血脂分析正常。外祖父母80岁时诊断糖尿病,体型匀称,平素口服二甲双胍控制血糖。母系亲属均无胰腺炎病史。
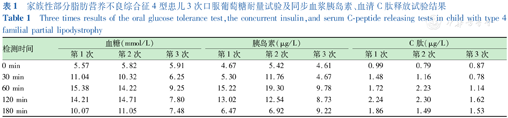
家族性部分脂肪营养不良综合征4型患儿3次口服葡萄糖耐量试验及同步血浆胰岛素、血清C肽释放试验结果
Three times results of the oral glucose tolerance test,the concurrent insulin,and serum C-peptide releasing tests in child with type 4 familial partial lipodystrophy
家族性部分脂肪营养不良综合征4型患儿3次口服葡萄糖耐量试验及同步血浆胰岛素、血清C肽释放试验结果
Three times results of the oral glucose tolerance test,the concurrent insulin,and serum C-peptide releasing tests in child with type 4 familial partial lipodystrophy
| 检测时间 | 血糖(mmol/L) | 胰岛素(μg/L) | C肽(μg/L) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 第1次 | 第2次 | 第3次 | 第1次 | 第2次 | 第3次 | 第1次 | 第2次 | 第3次 | |
| 0 min | 5.57 | 5.82 | 5.91 | 4.67 | 5.42 | 4.61 | 0.99 | 0.79 | 0.87 |
| 30 min | 11.04 | 10.32 | 6.25 | 5.30 | 11.76 | 4.67 | 1.48 | 1.16 | 0.78 |
| 60 min | 15.38 | 14.22 | 9.25 | 15.22 | 19.30 | 9.78 | 1.72 | 2.23 | 1.14 |
| 120 min | 14.21 | 14.71 | 7.80 | 13.02 | 12.54 | 8.73 | 2.24 | 2.30 | 1.62 |
| 180 min | 10.07 | 11.05 | 7.48 | 6.47 | 6.92 | 9.22 | 1.86 | 1.49 | 1.53 |
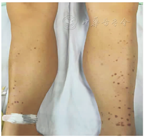
体格检查:血压114/70 mmHg(1 mmHg=0.133 kPa),身高170 cm,体重60 kg,体重指数(BMI) 20.8 kg/m2。皮下脂肪厚度:上臂4 mm、腹部11 mm、大腿7 mm。神志清,精神萎靡,嗜睡,皮肤干燥,颈部和腋下未见黑棘皮,无紫纹,躯干部及双下肢多发皮疹,粟粒样至黄豆粒大小,黄红色,部分中央部黄色颗粒大小物质,深大呼吸,双肺呼吸音清,心律齐,心音有力,腹软,肝脾肋下未及,下肢肌肉肥大(图1),四肢末梢凉。神经系统查体阴性。
辅助检查:血常规:白细胞19.71×109/L[正常范围:(4.00~10.00)×109/L]、中性粒细胞百分比75.60%(正常范围:50.00%~75.00%)、血红蛋白158 g/L(正常范围:110~140 g/L)、血小板365×109/L[正常范围:(85~303)×109/L];尿常规:葡萄糖(+++)、酮体(+++)、酸碱度5;尿淀粉酶985 μg/L(正常范围:0~450 μg/L);血淀粉酶82 U/L(正常范围:0~100 U/L);脂肪酶424.2 U/L(正常范围:13.0~63.0 U/L);C反应蛋白128.44 mg/L(正常范围:0~8.00 mg/L);血气分析及电解质:pH 7.08(正常范围:7.34~7.45)、实际碳酸氢根(AB)5.5 mmol/L(正常范围:21.4~27.3 mmol/L)、阴离子间隙30 mmol/L(正常范围:10~18 mmol/L)、渗透压294 mOsm/L(正常范围:270~300 mOsm/L)、钠137 mmol/L(正常范围:136~145 mmol/L)、钾4.5 mmol/L(正常范围:3.5~5.2 mmol/L);血脂分析:总胆固醇>检测值上限,三酰甘油>检测值上限;糖化血红蛋白15.3%(3.8%~6.0%);糖尿病自身抗体谱:阴性;口服葡萄糖耐量试验及同步血浆胰岛素、C肽释放试验结果:血糖(mmol/L):0 min 5.91、30 min 6.26、60 min 9.25、120 min 7.70、180 min 7.48,C肽(μg/L):0 min 0.87、30 min 0.78、60 min 1.14、120 min 1.62、180 min 1.53,胰岛素(μg/L):0 min 4.61、30 min 4.67、60 min 9.78、120 min 8.73、180 min 9.22;肝肾功能正常。腹部B超:肝右叶高回声团,胰尾周边组织回声欠均匀,左上腹腔近脾脏下方积液班网膜组织水肿;腹部CT:考虑胰腺炎。初步诊断:(1)急性胰腺炎;(2)DKA;(3)糖尿病。入院后,先后予禁食、低脂饮食及糖尿病饮食,奥美拉唑、生长抑素及胰岛素治疗后,胰腺炎好转,血糖平稳后出院。
本研究通过苏州大学附属儿童医院医学伦理委员会批准(批准文号:2021CS003),患儿监护人签署知情同意书。
经患儿及家属知情同意,分别抽取患儿、患儿父母、患儿妹妹外周静脉抗凝血3 mL于苏州大学附属儿童医院行全外显子基因及线粒体环基因检测,基因序列结果分析显示:患儿携带PLIN1 c.1325delG (p.G442Afs*99)杂合突变和SPINK1 c.194+2T>C(p.?)杂合突变,变异均来源于母亲,PLIN1 c.1325delG(p.G442Afs*99)目前尚未见文献报道,参照美国医学遗传学和基因组学学会指南评级证据(PVS1_Mo-derate + PM2_Supporting + PP4)[4],该变异被评级为致病变异。SPINK1 c.194+2T>C(p.?)目前已有文献报道,为中国慢性特发性胰腺炎患者中最常见的致病变异[5]。外祖父母因身体原因,无法获取血液标本。患者家系图及基因测序结果见图2。
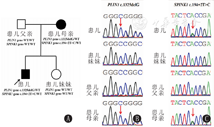
注:黑色箭头示患儿;红色箭头示突变位点 The black arrow indicates the child;red arrow indicates mutation site
患儿13岁5个月和14岁1个月时,因DKA分别第4次(2021年11月10日)、第5次(2022年7月3日)入院,入院前均有暴饮暴食病史,伴高脂血症,但是严格低脂饮食、胰岛素强化治疗后,DKA可快速纠正,高脂血症好转。目前患儿出院予严格饮食指导、胰岛素治疗,目前血糖控制良好,血脂正常,下肢黄色瘤较前消褪。患儿临床特点总结见表2。
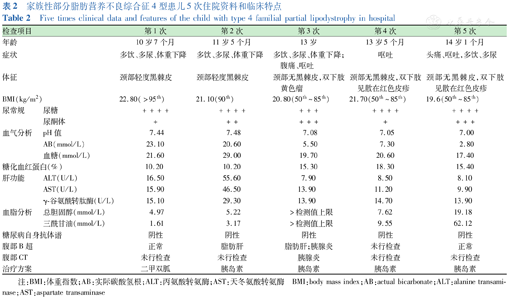
家族性部分脂肪营养不良综合征4型患儿5次住院资料和临床特点
Five times clinical data and features of the child with type 4 familial partial lipodystrophy in hospital
家族性部分脂肪营养不良综合征4型患儿5次住院资料和临床特点
Five times clinical data and features of the child with type 4 familial partial lipodystrophy in hospital
| 检查项目 | 第1次 | 第2次 | 第3次 | 第4次 | 第5次 | |
|---|---|---|---|---|---|---|
| 年龄 | 10岁7个月 | 11岁5个月 | 13岁 | 13岁5个月 | 14岁1个月 | |
| 症状 | 多饮、多尿、体重下降 | 多饮、多尿、体重下降 | 多饮、多尿、体重下降;腹痛、呕吐 | 呕吐 | 头痛、呕吐,多饮、多尿 | |
| 体征 | 颈部轻度黑棘皮 | 颈部轻度黑棘皮 | 颈部无黑棘皮,双下肢黄色瘤 | 颈部无黑棘皮,双下肢见散在红色皮疹 | 颈部无黑棘皮,双下肢见散在红色皮疹 | |
| BMI(kg/m2) | 22.80(>95th) | 21.10(90th) | 20.80(50th~85th) | 21.70(50th~85th) | 19.6(50th~85th) | |
| 尿常规 | 尿糖 | ++++ | ++++ | +++ | ++++ | ++++ |
| 尿酮体 | + | ++ | +++ | + | +++ | |
| 血气分析 | pH值 | 7.44 | 7.48 | 7.08 | 7.05 | 7.00 |
| AB(mmol/L) | 23.10 | 20.60 | 5.50 | 7.30 | 2.80 | |
| 血糖(mmol/L) | 21.60 | 29.00 | 19.70 | 20.60 | 17.40 | |
| 糖化血红蛋白(%) | 10.20 | 10.20 | 15.30 | 18.30 | 15.40 | |
| 肝功能 | ALT(U/L) | 16.50 | 55.60 | 7.90 | 8.50 | 8.10 |
| AST(U/L) | 15.90 | 46.50 | 13.90 | 11.20 | 9.90 | |
| γ-谷氨酰转肽酶(U/L) | 15.10 | 29.30 | 13.90 | 14.70 | 13.90 | |
| 血脂分析 | 总胆固醇(mmol/L) | 4.97 | 5.22 | >检测值上限 | 7.62 | 19.18 |
| 三酰甘油(mmol/L) | 1.61 | 3.17 | >检测值上限 | 9.55 | 62.12 | |
| 糖尿病自身抗体谱 | 阴性 | 阴性 | 阴性 | 阴性 | 阴性 | |
| 腹部B超 | 正常 | 脂肪肝 | 脂肪肝;胰腺炎 | 未行检查 | 正常 | |
| 腹部CT | 未行检查 | 未行检查 | 胰腺炎 | 未行检查 | 未行检查 | |
| 治疗方案 | 二甲双胍 | 胰岛素 | 胰岛素 | 胰岛素 | 胰岛素 | |
注:BMI:体重指数;AB:实际碳酸氢根;ALT:丙氨酸转氨酶;AST:天冬氨酸转氨酶 BMI:body mass index;AB:actual bicarbonate;ALT:alanine transaminase;AST:aspartate transaminase
Gandotra等[6]首次报道了3例以部分脂肪萎缩、严重血脂异常和胰岛素抵抗性糖尿病家系病例,目前共10个FPLD4家系病例被报道[7,8,9]。在已报道病例中,患者确诊年龄13~69岁,大部分患者在中年时期确诊,少数青少年患者因家庭成员确诊后被诊断,男女均有发病。患者有不同程度的脂肪萎缩,以四肢明显,可伴小腿肌肉肥大,同时多合并肥胖症、非酒精性脂肪性肝病、胰岛素抵抗性糖尿病及高三酰甘油血症等,成年女性患者多存在多毛及多囊卵巢综合征。在既往病例中,糖代谢异常多表现为胰岛素抵抗性糖尿病,患者在青年早期出现糖耐量异常,若不控制肥胖,随着BMI增高和年龄增长,逐渐发展成糖尿病,且胰岛素治疗效果不佳,但尚未见DKA报道。
FPLD4的发病机制与PLIN1基因突变有关,PLIN1定位于染色体15q26.1,含522个氨基酸,编码重要的脂滴包被蛋白perilipin 1,包含与自水解酶结构域蛋白5(ABHD5)结合的必需氨基酸区域(氨基酸382-429)、稳定ABHD5在脂滴定位的C末端(氨基酸398起)和可磷酸化位点(Ser492和Ser517)等重要结构,其主要表达在脂肪细胞和甾体生成细胞的脂滴表面,是脂滴代谢的关键性调控分子,具有双重作用[10,11,12]。目前已报道的5个PLIN1基因变异均为移码突变,分别为p.Val398Glyfs*166、p.Tyr401Leufs*165、p.Pro403Argfs*164、p.Leu404ALafs*158及p.Pro439Valfs*125[6,7,8,9]。前4种突变所致氨基酸改变均发生在perilipin 1蛋白C末端和ABHD5结合的必需氨基酸区域,这一改变使C末端蛋白延长,基础状态下ABHD5不能结合变异的perilipin 1并与脂滴分离,进而活化脂肪三酰甘油酶,增加三酰甘油分解,游离脂肪酸生成增加,故机体脂肪含量下降;多余的游离脂肪酸沉积在非脂肪细胞中并产生一定的脂毒性,如非酒精性脂肪肝和胰岛素抵抗的形成,同时,胰岛素抵抗使得胰岛素抑制激素敏感性脂肪酶作用减弱,加剧了游离脂肪酸的异位沉积[13],这与FPLD4的临床特点一致。p.Pro439Valfs*125突变位点位于ABHD5结合区域外,对结合ABHD5无影响,但C端蛋白延长,功能研究提示蛋白表达水平降低,基础脂肪分解率升高[7]。Gandotra等[6]和Kozusko等[7]研究发现,p.Val398Glyfs*166、p.Leu404ALafs*158及p.Pro439Valfs*125突变患者脂肪细胞中脂滴均变小。小的脂滴可提供更大的体表面积,从而使脂滴更快速地被水解[10]。本病例携带的变异p.Gly442Alafs*99发病机制可能与p.Pro439Valfs*125突变类似,突变位点发生在perilipin 1蛋白C末端,使C末端蛋白延长,影响perilipin 1蛋白功能,使脂肪分解异常。
本病例10岁起病,以糖尿病为首发临床表现,初诊时轻度肥胖伴轻度黑棘皮,无库欣貌,无血脂异常,病程中伴多次DKA发生,并逐渐出现脂代谢异常并发症,如高脂血症、非酒精性脂肪肝、黄色瘤及急性胰腺炎等,监测患儿胰岛β细胞功能提示呈进行性下降趋势。由于患儿起病年龄小,脂肪萎缩程度不明显,导致疾病诊断和糖尿病分型延迟。基因检测提示患儿存在PLIN1基因突变,结合患儿临床表现,目前FPLD4可诊断。基因检测中同时发现SPINK1 c.194+2T>C(p.?)变异,该变异在2000年被证实与慢性胰腺炎有关[14];SPINK1剪接突变体(c.194+2T>C,也称为IVS3+2T>C)是中国、日本和韩国最常见的致病突变[5,14,15],该失功能突变使胰酶抑制蛋白减少,胰蛋白酶活性增加,胰蛋白酶与其抑制蛋白的结合失衡,导致胰腺损伤,从而增加了胰腺炎发生的风险,尤其是在其他遗传因素和环境因素影响下[14,16]。故患儿病程中发生的急性胰腺炎,一方面与患儿平素饮食自控能力差、高脂饮食及高血糖有关,另一方面与患儿存在PLIN1突变和SPINK1突变有关。环境与基因的相互作用参与了患儿疾病的进展,整个病程中,患儿病情进展快,口服葡萄糖耐量试验及同步血浆胰岛素、C肽释放试验提示患儿胰岛素β细胞功能快速破坏,同时胰腺炎的发生可使胰岛β细胞丢失,导致内源性胰岛素分泌减少[17]。胰岛β细胞功能减退使患儿耐受高血糖的能力下降,这与患儿反复发生DKA有关,严重危害患儿身心健康。
患儿母亲携带与患儿相同的2个杂合变异,但在患儿母亲病程中仅糖尿病发生,且口服二甲双胍可控制血糖,无反复DKA、高血脂及胰腺炎发生,月经规律,无多毛,推测其原因可能与2人饮食习惯和生活方式差异有关。在Chen等[8]的报道中,1例15岁女性FPLD4患儿患有胰岛素抵抗性糖尿病,由于其平素低碳水、低脂饮食,坚持运动,因而无明显肥胖,脂肪萎缩表现不明显,口服二甲双胍治疗也可维持血糖平稳。故饮食、锻炼等生活方式的调整是预防和治疗糖脂代谢并发症的重要手段之一。
FPLD4的治疗以对症治疗为主,低脂、糖尿病饮食及适当运动是重要的辅助手段,胰岛素、胰岛素增敏剂(二甲双胍、噻唑烷二酮类等)和降脂药物应用可改善血糖、血脂紊乱所致代谢并发症。本病例已出现胰岛β细胞功能损伤,目前治疗以低脂、糖尿病饮食,健康运动,胰岛素皮下注射为主,同时需避免诱发胰腺炎的生活因素,定期进行胰腺功能的相关检查。
所有作者均声明不存在利益冲突

























