
发热伴血小板减少综合征(severe fever with thrombocytopenia syndrome, SFTS)是大别班达病毒(Dabie bandavirus, DBV)感染引起的一种以发热、白细胞和血小板减少、多器官损伤为临床特点的新发突发传染病。DBV导致的免疫系统紊乱是SFTS发病的重要机制之一。单核/巨噬细胞是固有免疫的重要成员,是DBV感染的靶细胞,其与病毒的相互作用在DBV致病过程中扮演重要角色。本文就DBV感染人体后单核/巨噬细胞介导的免疫效应特点及机制的相关研究进展进行综述。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
发热伴血小板减少综合征(severe fever with thrombocytopenia syndrome, SFTS)是由大别班达病毒(Dabie bandavirus, DBV)感染引起的一种新发急性传染病,主要由蜱虫叮咬传播,临床表现为发热、白细胞下降、血小板减少、消化道症状等,严重病例可出现多器官功能衰竭以及临床死亡。SFTS主要以散发形式在我国河南、山东、安徽、湖北、辽宁、浙江和江苏省等地区流行,病死率为6%~30%[1]。单核/巨噬细胞是机体非特异性免疫的主要组成部分,同时也是DBV感染的重要靶细胞之一,其在SFTS的发病机制中起着重要作用。本文就DBV感染机体后单核/巨噬细胞介导的免疫效应特点及机制的相关研究进展予以综述。
DBV属于白纤病毒科,班达病毒属,是一种单股负链RNA病毒,包括大(L)、中(M)、小(S)3个片段,L片段负责病毒RNA的复制和mRNA的合成,M片段裂解产生糖蛋白Gn/Gc,进而促进病毒进入靶细胞,S片段转录产生病毒核蛋白和非结构蛋白,保护其不被免疫识别[2,3]。2012年通过流行病学、实验室检查、病毒分离等一系列方法,首次阐明这种以发热、血小板减少、白细胞减少的疾病是由一种新型布尼亚病毒引起,并将其命名为SFTS布尼亚病毒[1]。2014年国际病毒分类委员会将这种新型病毒命名为发热伴血小板减少综合征布尼亚病毒(severe fever with thrombocytopenia syndrome bunyavirus, SFTSV),后来更名为Dabie bandavirus,并于2019年重新分类为白纤病毒科(Phenuiviridae),班达病毒属(Bandavirus)。
单核/巨噬细胞是人体非特异性免疫系统的重要组成部分。单核细胞由骨髓中的造血干细胞分化而来,成熟的单核细胞占外周血白细胞的10%,可在血液停留1~2 d或在集落因子的作用下,迁移到组织分化为巨噬细胞[4]。巨噬细胞除了由循环血液中的单核细胞转变而来,还可由胎儿时期肝脏的祖细胞、卵黄囊发育而来,不受血液中单核细胞影响,又称为组织巨噬细胞,如Kupffer细胞、肺泡巨噬细胞和小胶质细胞[5,6]。
单核/巨噬细胞是白细胞的一个亚群,是病毒入侵人体首要面对的"卫士"。病毒感染后,巨噬细胞可以发挥以下作用:(1)产生抗病毒物质Ⅰ型干扰素(IFN),诱导IFN刺激基因表达、抑制病毒复制,并激活NK细胞和T细胞[7];(2)除IFN外,巨噬细胞还可产生IL-1β和IL-6,激活丝裂原活化蛋白激酶/细胞外信号调节激酶(MAPK/ERK)通路,促进抗病毒活性,如在乙型肝炎病毒(hepatitis B virus,HBV)感染过程中,巨噬细胞可以诱导生成IFN、TNF-α等可溶性炎症介质直接抑制HBV复制,或释放IL-12、诱导NK细胞活化发挥间接的抗病毒作用[8];(3)巨噬细胞在识别病毒后,吞噬病原体,将病毒肽提呈给T淋巴细胞,诱导适应性免疫应答[9]。单核/巨噬细胞通过直接吞噬、释放IFN或可溶性炎性介质调控人体免疫网络,抵御微生物对机体的侵袭,维持内环境稳定。
单核/巨噬细胞是布尼亚病毒常见的感染靶细胞,如裂谷热病毒可以有效地感染单核细胞来源的巨噬细胞,影响巨噬细胞产生趋化因子[10]。单核/巨噬细胞也是DBV感染的重要靶细胞之一。既往研究显示从SFTS患者外周血分离的单核细胞中可以检测到DBV,且DBV病毒载量与单核细胞减少水平呈正相关,重症患者中单核细胞数量降低[11]。体外细胞试验显示,DBV在人源巨噬细胞THP-1中有更高的增殖水平,表达较高水平的病毒结构蛋白抗原,THP-1细胞在DBV感染后3~4 d会出现明显细胞病变,包括肿胀、圆缩或胞质空泡化、融合,以及破碎脱落和出现拉丝状[12],并且可以在巨噬细胞中形成病毒包涵体[13]。DBV感染动物模型试验也显示脾脏是DBV感染的靶器官,DBV可有效感染小鼠原代巨噬细胞[14]。近来单细胞测序技术研究从转录组学水平上证实了外周血单核细胞和B细胞是DBV藏匿的重要细胞[15]。由此,单核/巨噬细胞虽然是免疫系统重要的组成部分,但DBV可以感染单核/巨噬细胞,将其加以调节利用,藏匿其中,协助病毒的复制和传播。
以往研究表明其他布尼亚病毒感染宿主细胞主要通过以下几个步骤:(1)病毒糖蛋白与宿主细胞因子相互作用,例如树突状细胞特异性细胞间黏附分子-3-结合非整合素分子(dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin, DC-SIGN)、硫酸乙酰肝素、非肌肉肌球蛋白重链ⅡA;(2)病毒表面的Gc蛋白与细胞膜融合;(3)病毒核糖核蛋白复合物释放到细胞质中,进行病毒复制[16]。DBV可通过不同途径感染单核/巨噬细胞。病毒进入细胞需依赖细胞受体,DC-SIGN是一种表达在树突状细胞和巨噬细胞表面的Ⅱ型膜受体,它的C型凝集素区可以与病毒表面的甘露糖受体相互作用[17]。DBV的包膜蛋白Gn/Gc以DC-SIGN为受体入侵宿主细胞,增强对细胞的侵袭性,其中DBV对人单核细胞来源的树突状细胞感染力最强,对人单核细胞来源的巨噬细胞感染力最弱[18]。另一种受体-酪氨酸激酶受体,广泛存在细胞表面,参与细胞增殖、分化、趋化、迁移等生命活动,血小板来源生长因子受体β(platelet-derived growth factor receptor-like protein β,PDGFRβ)作为酪氨酸激酶受体家族的一员,参与DBV感染靶细胞,DBV可以通过PDGFRβ激活蛋白激酶B(AKT)通路内吞进入细胞[19]。另外,DBV还可以利用其他方式进入靶细胞,例如利用细胞膜上网状蛋白,形成内吞囊泡,进入细胞[20],也有动物实验表明,人单核细胞可以通过细胞间接触将DBV传染给HuPBL-NCG小鼠[21],传递"病毒性颗粒",介导病毒传播[22]。病毒糖蛋白(如DBV的Gn/Gc)对病毒侵袭靶细胞至关重要,病毒糖蛋白与多种宿主细胞因子结合促进病毒对靶细胞黏附,进入靶细胞并在细胞中组装病毒颗粒,完成复制。
不同于肾综合征出血热,重症SFTS患者外周血单核细胞明显下降,且单核细胞下降的比例与病毒载量呈正相关[11],DBV造成患者单核细胞减少的原因可能有:(1)DBV直接感染单核细胞诱导凋亡:单核细胞是DBV感染的靶细胞之一[15,23,24],DBV在感染早期感染单核细胞,并在细胞中复制,抑制单核细胞抗凋亡基因IL-6、CD40配体(CD40L)的表达,促进单核细胞凋亡[25];(2)DBV感染导致骨髓抑制:单核细胞主要由骨髓造血干细胞发育而来,骨髓是DBV感染的主要部位之一[26],一项研究表明SFTS患者的骨髓象的有核细胞增生减低或极度减低,粒细胞生成障碍[27],但造血干细胞或造血微环境是否受到DBV的影响尚鲜见报道,不过感染后的骨髓抑制仍然可能是SFTS患者单核细胞减少的重要原因;(3)过度消耗:血小板减少是SFTS患者的重要临床特征,最近研究表明巨噬细胞参与血小板减少机制,C57/BL6小鼠感染DBV后,脾脏中的巨噬细胞数量大量增加,参与清除与DBV黏附的血小板,使血小板减少[14],但这也同时消耗了外周血的单核细胞,可能是SFTS患者单核细胞减少的原因之一。
DBV感染不仅可导致单核细胞数量减少,还会引起单核细胞功能障碍。DBV感染可诱导患者的单核细胞亚群由经典型向中间型分化,中间型单核细胞诱导过度的IFN产生,且随着患者的病毒载量、疾病严重程度的加重,这种反应也更强烈[15,26]。此外,体外细胞研究显示DBV感染人源单核细胞THP-1后,通过上调miR-146b驱动巨噬细胞向M2表型分化,从而促进病毒脱落,并导致病毒传播[28],M2型巨噬细胞活化会导致IL-10过度分泌,产生低炎症状态,这种炎症反应并不受机体调控,过高的IL-10水平加重了SFTS患者的重症化风险[29]。另一项动物实验表明,DBV靶向作用于巨噬细胞的TLR2受体,产生IL-10,构建适合病毒复制的免疫抑制环境[30]。由单核细胞分化而来的髓样树突状细胞(myeloid dendritic cell,mDC)在被DBV感染后表现为CD86表达减少,影响了mDC的抗原提呈功能,从而进一步影响了T、B细胞的活化[25]。临床研究还发现SFTS患者外周血单核细胞虽然TLR4表达正常甚至升高,但在脂多糖刺激下产生TNF-α的水平显著低于健康人群,提示DBV感染导致单核细胞Toll样受体通路功能异常[11],表明Toll受体在DBV感染过程中发挥重要作用,或许可以作为未来的药物治疗靶点[30]。
细胞因子风暴造成的多器官功能损伤是SFTS患者重症化及临床死亡的重要原因,IL-6、IL-10、TNF-α、IFN-γ等炎性介质在SFTS患者中明显升高[31],巨噬细胞作为非特异免疫细胞是机体IL-1β、IL-6、IL-12和IL-23等炎症因子的主要来源细胞之一。DBV感染THP-1细胞可直接诱导TNF-α、IL-1β和IL-6生成[28],DBV的非结构蛋白可通过激活NF-κB诱导炎症因子释放[32],这是DBV促进细胞因子风暴形成、诱导机体损伤的重要途径。除了常见的炎症通路,近来发现,单核/巨噬细胞焦亡也参与DBV诱导的细胞因子风暴形成。焦亡是一种炎症细胞程序性死亡方式,主要通过各种炎症小体(如NLRP3)介导caspase半胱氨酸蛋白酶激活,造成细胞穿孔,胞质内容物流出,并伴随着大量促炎因子(IL-1β和IL-18)释放。研究显示,DBV感染THP-1细胞后可通过诱导BAK/BAX上调,使线粒体DNA释放到细胞质基质,激活焦亡,进一步的动物实验表明,焦亡引发的炎症能增加小鼠的病死率[33,34]。
铁蛋白的异常升高是SFTS患者,特别是重症患者的一个显著特征[35]。血清铁蛋白主要由肝脏产生,TNF-α和IL-6可以刺激血清铁蛋白升高,如前所述,DBV感染巨噬细胞可以促进TNF-α和IL-6的产生,从而使血清铁蛋白含量升高,使细胞产生氧化应激,造成损伤,铁蛋白也可以通过激活NF-κB,扩大炎症反应[36]。另外,SFTS患者过高的铁蛋白水平也是巨噬细胞活化综合征或又称继发性噬血细胞性淋巴组织细胞增生症(sHLH)的重要诊断标准之一。临床研究发现SFTS死亡患者铁蛋白水平显著上升,尸检可在肝脏、脾脏等多器官中见到巨噬细胞浸润[37],对SFTS患者进行骨髓穿刺发现,有噬血现象者占78.1%,噬血特点以巨噬细胞吞噬红细胞和血小板最常见(60%),同时还可看到巨噬细胞吞噬中性粒细胞、细胞碎片并伴有巨噬细胞成熟障碍[27,38,39]。尽管sHLH多以散发为主,但Kim等[40]认为,噬血现象可能是SFTS患者普遍存在的骨髓特点,sHLH的发生也促进SFTS患者血细胞进一步下降。
DBV感染导致铁蛋白水平升高的具体机制尚未完全明确,单核/巨噬细胞铁死亡可能参与其中。铁死亡是近来发现的一种与焦亡相似的炎症细胞死亡方式,主要表现为脂质过氧化。细胞的主要离子转运蛋白SLC11A2是铁死亡通路的一个关键基因,DBV感染THP-1细胞后,转录组学研究结果发现SLC11A2水平明显升高,表明单核/巨噬细胞铁死亡通路也可能在DBV感染过程中过度激活[41],然而此机制还需要更多的实验来证实。上述研究表明,DBV感染过程中单核/巨噬细胞通过多种方式释放大量炎性介质导致预后不佳,DBV感染不仅能直接促进巨噬细胞释放IL-6等炎症因子,亦能诱导细胞死亡,通过焦亡、铁死亡的方式进一步扩大炎症效应,过高的铁蛋白水平可以继发噬血细胞综合征加重患者病情。IL-6、IL-10、铁蛋白水平可以用作重症患者的早期预警指标,糖皮质激素可以拮抗IL-1、IL-6等炎症因子的合成和分泌,但近期的两项研究表明,早期使用糖皮质激素不能明显改善DBV感染患者的生存率[42,43]。DBV感染形成细胞因子风暴机制复杂,单核/巨噬细胞在其中发挥重要作用。
IFN是一种强大的抗病毒介质,维甲酸诱导基因Ⅰ(RIG-Ⅰ)属于RIG-Ⅰ样受体(RLRs)家族,参与识别病毒RNA并介导Ⅰ型IFN参与的免疫应答。RIG-Ⅰ可以识别病毒RNA产物激活免疫系统,DBV感染人体后,DBV的非结构蛋白可被RIG-Ⅰ和Toll样受体识别,激活下游髓样分化因子88(MyD88)和IFN-β启动子刺激因子1(IPS-1,又称为MAVS),诱导巨噬细胞产生IFN和炎症因子,抑制病毒复制[44]。然而,病毒为了能正常完成转录、蛋白质合成、病毒组装和释放过程,进化出一套免疫逃逸机制。RIG-Ⅰ受体识别DBV后,病毒的非结构蛋白将TRIM25捕捉入包涵体,阻碍RIG-Ⅰ的激活,减少IFN的产生[45],DBV不仅可以避免被宿主细胞识别,也可以抑制IFN的传导,STAT是Ⅰ型IFN的重要激活位点,DBV的非结构蛋白作用于STAT1的S727位点,抑制其磷酸化,从而阻断IFN信号传导。不同于上述DBV对炎症因子生成的促进作用,DBV的非结构蛋白也可以通过抑制NF-κB信号通路,抑制IFN-β信号传导[23]。而最新的研究认为,DBV的非结构蛋白主要影响STAT2的磷酸化和核移位,通过阻断JAK-STAT途径抑制IFN信号的传导,与STAT1作用后并不影响Ⅰ型IFN的传导[46]。而多项研究表明,无论DBV通过哪种方式影响IFN通路,IFN的信号传导在DBV感染中都出现抑制,例如一项通过对外周血单核细胞相关基因的分析发现,随着疾病的严重程度增加,单核细胞中的TLR3出现下调,IFN调节因子(IRF3、IRF7)也出现了相同的变化趋势,IFN-α和IFN-β在死亡患者中明显下调,这提示IFN在DBV感染中应答不佳,而TLR3通路损伤参与了IFN的应答不佳[47],IFN的应答不佳,可能会加重DBV感染的严重程度。动物实验表明,阻断IFN-α受体会增强小鼠对DBV的易感性,增加了小鼠的病死率[44,48]。DBV通过以上各种方式抑制IFN的产生及后续免疫系统激活,从而达到免疫逃逸的目的。
SFTS作为一种新发现的传染病,病死率高,目前仍缺少有效的抗病毒药物及预防疫苗。本文总结了DBV感染与单核/巨噬细胞相关的临床及基础研究进展,提示单核/巨噬细胞作为DBV感染的重要靶细胞,既是介导机体抗病毒固有免疫系统的重要组成部分,也在细胞因子过度产生导致炎症损伤中发挥重要作用。此外,单核/巨噬细胞还被病毒利用作为逃逸免疫的工具,一方面DBV在感染早期通过多种方式损伤单核/巨噬细胞,使单核/巨噬细胞数量减少,从而实现免疫逃逸、完成病毒复制过程;另一方面DBV通过Toll样受体激活单核细胞,释放大量炎症因子,诱导细胞发生铁死亡或焦亡加重炎症反应、损伤组织细胞(图1)。因此,阐明单核/巨噬细胞在DBV致病过程中的作用,有助于揭示SFTS的发病机制,为未来发展新的免疫治疗方式提供帮助。
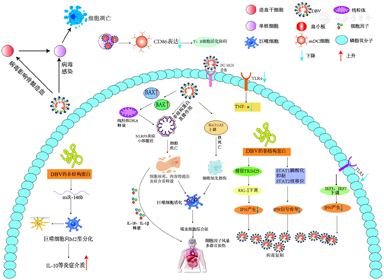
注:DBV:大别班达病毒;mDC:髓样树突状细胞;DC-SIGN:树突状细胞特异性细胞间黏附分子-3-结合非整合素分子;TLR:Toll样受体;RIG-Ⅰ:维甲酸诱导基因Ⅰ;IRF:IFN调节因子
刘亚男,郑昕.单核/巨噬细胞在发热伴血小板减少综合征中的研究进展[J].中华微生物学和免疫学杂志,2024, 44(1): 27-33. DOI: 10.3760/cma.j.cn112309-20230408-00086.
所有作者声明无利益冲突

























