
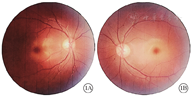
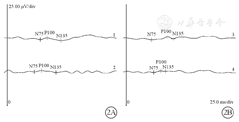
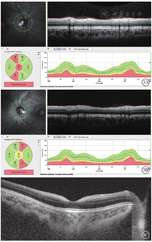
患者男,15岁。因走路易跌倒3年至北京儿童医院神经内科就诊,因双眼进行性视力下降转诊于眼科。患儿1个月前无发热性抽搐2次,表现为双眼上翻,口唇发绀,牙关紧闭,约1~2 min缓解。无全身病史及家族遗传病史。眼科检查:右眼、左眼最佳矫正视力均为20/100;屈光状态:右眼-4.50 DS/-2.75 DC×180°、左眼-2.00 DS/-2.75 DC×5°。眼压:右眼、左眼分别为19、16 mm Hg(1 mm Hg=0.133 kPa)。双眼眼球运动正常;瞳孔圆,直径2.5 mm,对光反射存在;晶状体可见少量白色点状混浊。眼底检查,双眼视盘颜色淡、边界清楚,杯盘比约0.3~0.4,黄斑呈"樱桃红斑"改变(图1)。视觉诱发电位(VEP)检查,双眼在低、中、高空间频率刺激下P100波振幅明显降低(图2)。视网膜电图(ERG)检查,双眼分别在明、暗适应刺激下a、b波振幅大致正常。光相干断层扫描(OCT)检查,双眼视盘周围神经纤维层厚度降低,右眼、左眼分别为76、79 μm;双眼黄斑中心凹形态大致正常,神经节细胞层因唾液酸沉积反射增强,与神经纤维层反射强度相同,界限不清,融合为一层,两层厚度正常(图3)。神经系统检查,四肢肌力及肌张力正常,腱反射正常,双侧巴宾斯基征(+),踝阵挛(-)。共济运动失调检测,指鼻试验不准,走直线不稳。颅脑核磁共振成像(MRI)检查及脑电图检查未见异常。眼科初步诊断:双眼视神经病变、双眼黄斑病变。神经内科建议进一步完善外周血基因检查明确诊断。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
患者男,15岁。因走路易跌倒3年至北京儿童医院神经内科就诊,因双眼进行性视力下降转诊于眼科。患儿1个月前无发热性抽搐2次,表现为双眼上翻,口唇发绀,牙关紧闭,约1~2 min缓解。无全身病史及家族遗传病史。眼科检查:右眼、左眼最佳矫正视力均为20/100;屈光状态:右眼-4.50 DS/-2.75 DC×180°、左眼-2.00 DS/-2.75 DC×5°。眼压:右眼、左眼分别为19、16 mm Hg(1 mm Hg=0.133 kPa)。双眼眼球运动正常;瞳孔圆,直径2.5 mm,对光反射存在;晶状体可见少量白色点状混浊。眼底检查,双眼视盘颜色淡、边界清楚,杯盘比约0.3~0.4,黄斑呈"樱桃红斑"改变(图1)。视觉诱发电位(VEP)检查,双眼在低、中、高空间频率刺激下P100波振幅明显降低(图2)。视网膜电图(ERG)检查,双眼分别在明、暗适应刺激下a、b波振幅大致正常。光相干断层扫描(OCT)检查,双眼视盘周围神经纤维层厚度降低,右眼、左眼分别为76、79 μm;双眼黄斑中心凹形态大致正常,神经节细胞层因唾液酸沉积反射增强,与神经纤维层反射强度相同,界限不清,融合为一层,两层厚度正常(图3)。神经系统检查,四肢肌力及肌张力正常,腱反射正常,双侧巴宾斯基征(+),踝阵挛(-)。共济运动失调检测,指鼻试验不准,走直线不稳。颅脑核磁共振成像(MRI)检查及脑电图检查未见异常。眼科初步诊断:双眼视神经病变、双眼黄斑病变。神经内科建议进一步完善外周血基因检查明确诊断。
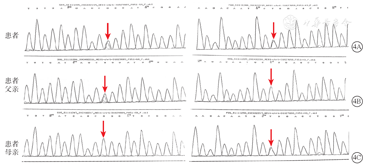
取得患者及其监护人知情同意后,取患者及其父母外周血行基因检测。结果显示,患者存在NEU1基因chr6:31829889 exon2 c.239C>T(p.P80L)杂合突变(来源于父亲)、NEU1基因chr6:31827858 exon5 c.982G>A(p.G328S)杂合突变(来源于母亲)(图4)。根据临床表现、黄斑部"樱桃红斑"改变及基因检测结果,最终诊断:Ⅰ型唾液酸沉积症(SD)。
SD是一种极为罕见的常染色体隐性遗传的神经代谢类疾病。它系NEU1基因突变导致α-N-乙酰神经氨酸酶(即唾液酸酶)异常,糖蛋白和糖脂的正常代谢分解受阻,导致唾液酸寡糖在细胞内异常蓄积所致。根据发病年龄和临床症状SD分为两型:Ⅰ型SD发病率为1/250 000~2 000 000成活婴儿,发病年龄多在10岁之后[1]。初期症状较轻,随着病情发展出现中枢神经系统表现,以肌阵挛最常见,另可伴有共济失调(88.3%)和癫痫(72.5%)。颅脑MRI多表现为正常[2]。临床上因其缺乏特异性表现常常造成误诊或漏诊。眼部在早期即可出现视力下降,发生率高达68.4%[2]。黄斑"樱桃红斑"具有明显的特异性,为诊断此病提供了重要线索。Ⅱ型SD于患儿出生即发病,表现为面部粗糙、肝胆脾肿大、骨骼异常、脊柱畸形及严重发育迟缓,常常在1岁内死亡[3,4]。本例患者最初症状表现为走路不稳,逐步进行性加重的肌阵挛和癫痫发作,与SD特征性肌阵挛、癫痫发作、小脑共济失调表现相一致,因此此类患者常首诊于神经内科。因患者出现视力进行性下降,经眼科检查发现眼底特征性黄斑"樱桃红斑"表现,特别注意的是我们发现其OCT显示唾液酸化代谢产物沉积在黄斑周围的神经节细胞层,导致其反射增强与神经纤维层界限不清。VEP检查提示双眼传导通路异常。基因检测结果发现,本例患者携带NEU1基因c.239C>T和c.982G>A复合杂合突变,分别来自表型正常的父母。结合以上信息,该患儿符合Ⅰ型SD诊断。
目前国内有关儿童SD鲜见报道。吕瑞娟等[5]纳入77例全球范围内发表的Ⅰ型SD患者数据进行分析,发现中国内地Ⅰ型SD患者的发病年龄明显偏小,为(10.8±2.7)岁;视力损害的发生率较其他亚洲地区更高,约81.8%~100.0%;39/72(54.2%)的患者存在黄斑"樱桃红斑"。"樱桃红斑"在临床上具有辨识度和特异性,既往研究报道Ⅰ型SD患者的"樱桃红斑"发生率在50%以上[6,7]。Ahn等[8]认为,发病早的患者较发病晚的患者,更容易出现"樱桃红斑"。SD患者出现"樱桃红斑"的可能机制是,唾液酸化代谢产物沉积在黄斑周围的神经节细胞层中,导致黄斑中心凹周围神经节细胞层透明度下降而呈现白色外观,中心凹缺乏神经节细胞而透出脉络膜红色,在白色背景的映衬下呈"樱桃红斑"的改变[9]。黄斑部神经节细胞最多,呈3~4层排列,随着大量的唾液酸寡糖或糖肽的代谢产物沉积,在OCT图像上表现为反射增强至与神经纤维层相同,两层间界线模糊、融合为一层;同时造成视神经逐步萎缩,导致视力损伤[10]。本例患者黄斑部神经节细胞层呈乳白色、在OCT上表现为反射增强与神经纤维层相同,给唾液酸代谢物沉积所致的发病机制提供了临床支持。
黄斑"樱桃红斑"还可见于其他溶酶体沉积症疾病,如神经节苷脂沉积症、神经元蜡样脂褐质沉积症、戈谢病、黏多糖病等。但这些疾病都有各自特点,临床上较容易鉴别[11]。如婴幼儿神经节苷脂沉积症发病较早,多在出生后6~7个月出现发育倒退,伴有表情淡漠、对声音刺激的"惊跳"现象、抽搐以及肌力改变等[12]。神经元蜡样脂褐质沉积症以进行性痴呆、发育倒退、癫痫、视力丧失、神经元内蜡样脂褐素贮积为表现。戈谢病患儿多表现为明显生长发育落后、骨骼受累、脾脏肿大、脾功能亢进血小板减少、贫血及眼球运动异常等。黏多糖病以面容粗陋、肝脾肿大和多发性骨发育不良为特征表现,伴或不伴进行性中枢神经系统疾病异常。因此,"樱桃红斑"对临床上诊断SD具有较高的特异性,是确诊SD的重要线索。
本例SD患者双眼VEP检查显示P100波振幅明显降低。Fan等[2]发现,几乎所有的Ⅰ型SD患者均可见潜时延长和巨大的体感诱发电位(SSEP),即使在没有视觉症状的SD患者中,VEP和SSEP也可能是Ⅰ型SD患者视力受损敏感的神经电生理标记物。Lai等[13]报道的17例Ⅰ型SD患者中,仅4例(23.5%)有视觉损害的临床症状,而16例(94.1%)发现伴有VEP异常表现,因此VEP检查对该病的视力损害有早期诊断价值,尤其适用于无法配合视力、OCT或视野检查的低龄儿童。
本例患者同时伴有癫痫、肌阵挛、共济失调等神经系统症状,尽管就诊时颅脑MRI检查及ERG早期无明显异常。出现癫痫的SD患者脑电图常显多棘波和多棘-慢波,随着病情进展,颅脑MRI检查显示大脑皮质萎缩或小脑萎缩[11, 14,15]。Lu等[15]通过Ⅰ型SD患者颅脑MRI检查发现,颅脑后部皮质受损最严重位于枕叶和颞叶,而患者出现大脑微结构损伤最初发生在后部视觉区域,随病情加重向更广泛的大脑功能区域扩展。病理学研究发现,在Ⅰ型SD患者的大脑中枢神经元内积累的唾液酸寡糖[16],癫痫发作可能提示病变范围增加或病情加重。
所有作者均声明不存在利益冲突

























