
分析苗勒管永存综合征(persistent Müllerian duct syndrome,PMDS)的临床特征,并探讨PMDS患儿的睾丸功能评估以及肿瘤的发生风险。
收集2010年1月至2022年4月于深圳市儿童医院住院诊断为PMDS的5例男患儿的临床资料,患儿中位年龄15个月,年龄范围为5~26个月。采用外生殖器男性化程度评分系统(external masculinization scores,EMS)进行评分,人绒毛膜促性腺激素激发试验进行性腺功能评估,行超声检查、遗传学基因检测、外科手术及病理学检查。
5例中有3例为双侧隐睾、1例为单侧隐睾、1例为睾丸横过异位。根据EMS评分,5例患儿外生殖器呈轻至中度男性化不全。4例患儿行人绒毛膜促性腺激素激发试验提示睾丸间质细胞功能良好(睾酮14.68~29.10 nmol/L),3例患儿抗苗勒管激素(anti-Müllerian hormone,AMH)低于年龄正常参考值,1例水平正常,1例初诊时未检测AMH。经盆腔超声检查发现苗勒管残留物(Müllerian remnants,MRs)3例,1例未行盆腔超声检查。5例患儿染色体核型分析均为46,XY。4例患儿全外显子测序结果为2例AMH基因变异、2例AMHR2基因变异。5例患儿均行双侧睾丸下降固定术和腹腔镜探查,均发现MRs。4例患儿行性腺病理均为发育不良的睾丸。
在睾丸下降不全患儿中应注意识别PMDS,并尽早行隐睾下降固定。PMDS发生睾丸肿瘤的风险远远高于其他隐睾患儿,需严密监测。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
苗勒管永存综合征(persistent Müllerian duct syndrome,PMDS)是一种罕见的46,XY性发育障碍(disorders of sex development,DSD),属常染色体隐性遗传性疾病,表现为轻度男性化不全、体内同时具有苗勒管残留物(Müllerian remnants,MRs),如发育不良的子宫、输卵管和阴道上三分之一段。根据致病基因不同分为PMDSⅠ型和PMDSⅡ型,分别由抗苗勒管激素(anti-Müllerian hormone, AMH)基因和抗苗勒管激素受体2(AMHR2)基因变异所致。自1939年Nilson[1]首次描述PMDS以来,其有报道病例约300例,既往研究大多数关注于PMDS的临床分型、MRs的解剖结构及外科手术处理等方面,罕有对睾丸功能(激素水平和病理)的评估。本研究总结了5例PMDS患儿的临床表现、性腺功能评估、超声结果、遗传学结果、性腺病理,同时进行随访,分析了PMDS的临床特征,并重点讨论了PMDS患儿的睾丸功能评估结果和影响因素以及肿瘤的发生风险。
收集2010年1月至2022年4月于深圳市儿童医院住院诊断为PMDS的5例男患儿的临床资料,患儿中位年龄15个月,年龄范围为5~26个月。针对5例PMDS患儿的临床表现、性腺功能评估、超声检查、遗传学结果、外科手术及性腺病理结果等资料进行分析。
本研究已通过深圳市儿童医院审核批准(2021097),所有参与本研究的患儿法定监护人均知情同意。
评价患儿外生殖器外观:完全女性表型为0分,正常男性表型为满分12分,小于7分为轻度男性化不全。
性激素、血清AMH、抑制素B(inhibin B,INH-B)等检查,并行促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)激发试验和人绒毛膜促性腺激素(human chorionic gonadotropin,hCG)激发试验[3]。hCG激发试验后睾酮>10 nmol/L认为睾丸间质细胞功能良好,AMH及INH-B位于参考范围内(按年龄段划分)认为存在睾丸支持细胞,AMH显著低于参考值范围认为睾丸支持细胞功能差,同时可能存在MRs[4]。
采用GE logic E8超声诊断仪,凸阵探头,频率3.5~12.0 MHz,线阵探头,频率9~12 MHz。所有患儿均在安静状态下进行检查,检查部位包括盆腔、腹股沟及阴囊,以评估性腺及MRs。
征得患儿监护人知情同意后,例2~5患儿分别采集EDTA抗凝外周血2 ml,提取基因组DNA检测,例2~4患儿行全外显子测序(whole exome sequencing, WES),例5患儿行定点变异位点验证。采用Sanger测序验证阳性基因变异位点,同时对患儿父母相应基因位点进行检测。
按照2015年美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)指南[5]判断患儿的基因变异分类。
根据术前DSD多学科协作组讨论结果,征得监护人同意后,对患儿行睾丸下降固定术、腹腔镜探查及性腺活检。睾丸病理学检查采用苏木精-伊红染色的石蜡包埋切片,同时进行免疫组织化学染色。免疫标志物包括:胚胎干细胞相关蛋白3/4(Oct3/4),原癌基因c-Kit蛋白(CD117),胎盘碱性磷酸酶(placental alkaline phosphatase,PLAP),Y染色体基因编码的睾丸特异性蛋白1(TSPY1)。
仅例2患儿在57个月时进行了门诊复查,包括AMH、INH-B、肿瘤标志物及盆腔腹股沟超声检查。
5例均为男患儿,均以"阴囊空虚"就诊,其中3例为双侧隐睾,1例为单侧隐睾,1例为睾丸横过异位(例5患儿),其在8月龄时手术,56月龄时因同胞(例4)确诊后进一步评估。(表1)

苗勒管永存综合征5例男患儿的临床症状及体征
苗勒管永存综合征5例男患儿的临床症状及体征
| 编号 | 初诊年龄(月) | 主诉 | 第二性征 | EMS评分 |
|---|---|---|---|---|
| 1 | 26 | 双侧阴囊空虚 | 双侧乳房B1期,腋毛A1期,阴毛PH1期,阴囊融合,双侧睾丸未触及,阴茎长2.5 cm(静息)和4.5 cm(勃起),尿道开口正常 | 6 |
| 2 | 10 | 右侧阴囊空虚,左侧腹股沟疝 | 双侧乳房B1期,腋毛A1期,阴毛PH1期,阴囊融合,右侧睾丸未触及,左侧睾丸位于阴囊内、活动度大,阴茎2.5 cm×1.0 cm(静息),尿道开口正常 | 10.5 |
| 3 | 15 | 双侧阴囊空虚,左侧腹股沟疝 | 双侧乳房B1期,腋毛A1期,阴毛PH1期,阴囊融合,右侧睾丸未触及,左侧睾丸位于腹股沟区,阴茎3.0 cm×1.3 cm(静息),尿道开口正常 | 10 |
| 4 | 16 | 双侧阴囊空虚,双侧腹股沟疝 | 双侧乳房B1期,腋毛A1期,阴毛PH1期,阴囊融合,双侧睾丸分别位于相应腹股沟区,阴茎2.8 cm×1.0 cm(静息),尿道开口正常 | 11 |
| 5 | 5 | 左侧阴囊空虚 | 双侧乳房B1期,腋毛A1期,阴毛PH1期,阴囊融合,左侧阴囊空虚,右侧阴囊可触及2个睾丸,阴茎发育可,尿道开口正常 | 12 |
注:EMS,外生殖器男性化程度评分系统;阴茎大小表示为"长度×横径"
常规生化、肿瘤标志物、肾上腺激素等检查均正常。
性腺功能评估情况:例1~4患儿行hCG激发试验提示睾丸间质细胞功能良好,睾酮水平为14.68~29.10 nmol/L;例1~3患儿AMH水平低下,例4患儿AMH水平位于正常参考值范围,例5患儿初诊时未检测AMH,在56月龄时,其AMH水平已明显低于正常参考值下限。(表2)
苗勒管永存综合征5例男患儿性腺功能评估
苗勒管永存综合征5例男患儿性腺功能评估
| 编号 | 检查年龄(月) | 促黄体生成素(U/L) | 卵泡刺激素(U/L) | 睾酮(nmol/L) | 双氢睾酮(pg/ml) | 抗苗勒管激素(ng/ml)a | 抑制素B(pg/ml)b | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 基础值 | 峰值 | 基础值 | 峰值 | 基础值 | hCG激发后 | 基础值 | hCG激发后 | ||||
| 1 | 26 | 0.28 | 1.62 | 0.73 | 3.68 | 0 | 14.68 | 21.97 | 381.14 | <1.0 | 207.944 |
| 2 | 10 | 1.82 | 5.97 | 2.16 | 5.62 | 0.06 | 15.13 | 16.95 | 142.14 | 12.2 | 164.59 |
| 3 | 15 | 0.06 | 2.69 | 0.21 | 3.48 | 0.09 | 26.43 | 23.78 | 138.26 | <0.01 | 224.88 |
| 4 | 19 | 0.14 | 4.81 | 2.07 | 14.79 | 0.16 | 29.1 | 62.31 | 169.59 | 134.4 | 146.94 |
| 5 | 56 | <0.12 | - | 1.39 | - | 0.17 | - | - | - | 20.55 | 386.96 |
注:a正常参考范围:6月龄~1岁为186.81(30.68~375.51)ng/ml,1~2岁为156.76(4.98~365.22)ng/ml,2~3岁为148.41(8.90~295.99)ng/ml,4~5岁为125.49(37.88~298.52)ng/ml;b正常参考值范围:6月龄~1岁217.59(33.17~418.50)pg/ml,1~2岁为163.23(33.78~339.64)pg/ml,2~3岁为133.38(18.90~293.13)pg/ml,4~5岁为79.08(31.03~225.78)pg/ml
例1~3可见MRs,均为发育不良的子宫;例4未发现MRs;例5未进行盆腔超声检查。例1~4患儿的性腺位于盆腔,例5患儿的性腺位于阴囊内,所有性腺均符合睾丸声像,无占位。其中2例患儿存在睾丸横过异位:例1为右侧睾丸横过异位至左侧盆腔、例5为左侧睾丸横过异位至右侧阴囊内。
5例患儿染色体核型分析均为46,XY。WES结果显示例2和例3的AMH基因变异、例4和例5的AMHR2基因变异,根据2015年ACMG指南,均为致病和可能致病的复合杂合变异,且与临床表型相符。
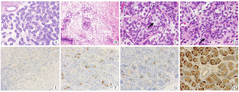
5例患儿均行双侧睾丸下降固定术和腹腔镜探查,均发现MRs(其中例4盆腔超声未发现MRs)(表3)。例1~4性腺病理诊断均为发育不良的睾丸,表现为光镜下可见睾丸间质疏松、纤维增生、水肿,曲细精管变形,生殖细胞成熟延迟(图1)。其中,例2患儿13月龄时睾丸苏木素-伊红染色为睾丸肿瘤细胞融合成细胞巢,免疫组织化学结果为:Oct3/4,个别曲细精管中心部(+),个别曲细精管基底部(+);CD117,个别曲细精管细胞(+);PLAP,散在曲细精管内个别细胞(+);TSPY1,部分曲细精管基底部(+)。病理诊断为双侧睾丸生殖细胞成熟延迟及灶状前原位生殖细胞瘤(图1)。
苗勒管永存综合征5例男患儿外科手术及性腺病理结果
苗勒管永存综合征5例男患儿外科手术及性腺病理结果
| 编号 | 手术年龄(月) | 睾丸下降固定术 | MRs | MRs切除 | MRs肿瘤 | (双侧)性腺病理 | 性腺肿瘤 |
|---|---|---|---|---|---|---|---|
| 1 | 26 | 是 | 存在发育不良的输卵管、子宫及阴道 | 部分切除 | 无 | 发育不良的睾丸 | 无 |
| 2 | 13 | 是 | 存在发育不良的输卵管及子宫 | 否 | 无 | 发育不良的睾丸 | 灶状前原位生殖细胞瘤(双侧睾丸) |
| 3 | 18 | 是 | 存在发育不良的输卵管及子宫 | 否 | 无 | 发育不良的睾丸 | 无 |
| 4 | 16 | 是 | 存在发育不良的输卵管及子宫 | 否 | 无 | 发育不良的睾丸 | 无 |
| 5 | 8 | 是 | 存在发育不良的输卵管及子宫 | 否 | 无 | 未做 | 无 |
注:MRs,苗勒管残留物
例2患儿57个月时AMH为2.23 ng/ml(正常参考范围37.88~298.52 ng/ml,平均值125.49 ng/ml),INH-B为155.48 pg/ml(正常参考范围31.03~225.78 pg/ml,平均值79.08 pg/ml),甲胎蛋白、β-hCG均阴性,超声显示双侧睾丸及MRs均无占位病变。
本研究在国内首次详细评估了PMDS患儿的睾丸功能(包括激素水平和病理结果),分析了其可能的影响因素,小结了PMDS患儿睾丸肿瘤的发生风险。PMDS患儿睾丸早在中位年龄17个月(13~26个月)时血中激素指标尚在正常范围时,病理已出现睾丸发育不良的表现。术后随访,PMDS患儿AMH水平下降幅度大于健康男童,即使早期进行了隐睾下降固定术,仍应警惕睾丸功能损害进展。4例完成睾丸活检的患儿中,有1例在13月龄时出现双侧睾丸灶状前原位生殖细胞瘤,长期随访肿瘤性质暂未进展。
PMDS可分为3种类型:腹股沟子宫疝型占20%~30%,睾丸横过异位型(transverse testicular ectopic,TTE)占10%,双侧高位隐睾型占60%~70%[6]。腹股沟子宫疝型和TTE对PMDS的诊断特异性较高,相对易于识别。最常见的双侧高位隐睾型患儿,若未行盆腔影像学检查,易漏诊,甚至经过盆腔超声也有可能漏诊。Tian等[7]报道了12例PMDS患儿,其中仅7例在术前超声检查识别。因此,还需在腹腔镜探查时注意寻找是否存在MRs。Shalaby等[8]统计了2009年1月至2013年8月埃及阿西乌特大学附属医院泌尿外科因睾丸下降不全就诊的832例患儿,经腹腔镜探查后PMDS的诊断率为2.3%(19/832),迄今报道的例数明显低估了发病率。
约88%的PMDS由AMH和AMHR2基因发生变异所致[9],其他可能参与的通路还有SMAD、WNT/β-连环蛋白、SF1、GATA1、HSP70、WT1、GATA4[10,11,12,13]。男患儿中,AMH除了熟悉的诱导胎儿苗勒管退化外,还参与了诱导睾丸下降,调控间质、支持及生殖细胞功能。Behringer等[14]在10周龄以上AMH基因纯合缺陷的雄鼠睾丸活检中发现其睾丸间质细胞增生。Mishina等[15]建立了1.5~11.0月龄大的AMHR2基因纯合缺陷雄鼠模型,其睾丸病理表现为:睾丸支持细胞萎缩、空泡变性,睾丸间质细胞增生,生精小管变性钙化、无精子生成等。迄今为止,在PMDS患儿中对睾丸功能和病理评估的报道较少。Saleem等[16]总结了27例年龄在0.2~19.0岁的巴基斯坦PMDS患儿,2例评估了性腺功能(仅测定睾酮水平),进行睾丸活检的9例患儿其结果为睾丸发育不良和不同程度的成熟延迟。对PMDS的生育调查显示96.7%(145/150)无法生育,精液分析为弱精、少精甚至无精[17,18,19,20]。本研究中,5例患儿进行异位睾丸下降固定术的中位年龄为16个月(8~26个月),但性腺活检均已提示为发育不良的睾丸(临床激素水平尚处于年龄参考范围内);例2和例5患儿尽管16月龄内已行异位睾丸下降固定术,但仍早在4~5岁时出现AMH水平显著低下。因此,一方面在睾丸下降不全患儿中应注意识别诊断PMDS,并尽早行隐睾下降固定;另一方面,结合睾丸病理及临床激素变化,可能存在睾丸位置以外的其他因素影响了PMDS的睾丸功能,需进一步明确病因方可指导预后。此外,PMDS患儿隐睾下降固定术后仍应长期随访睾丸功能,必要时行精液或精子冻存。
PMDS发生睾丸肿瘤的风险类似于腹腔型隐睾患儿,而高于腹股沟型隐睾患儿。国内报道腹腔内隐睾恶变率为22.7%,腹股沟型隐睾恶变率为6.8%[21]。Bucci等[22]报道PMDS患儿睾丸肿瘤的发生率可高达18%,多为青春期后发生。Picard等[9]统计了18岁以上PMDS患儿发生睾丸肿瘤的风险为33%,高于其他类型XY,DSD。PMDS的睾丸肿瘤以精原细胞瘤最常见,其他为胚胎癌、畸胎瘤、绒癌、性母细胞瘤、卵黄囊瘤、混合性生殖细胞肿瘤和原位生殖细胞瘤变(germ cell neoplasia in situ,GCNIS)[23]。Manassero等[24]综述了37例发生睾丸肿瘤的PMDS患儿,6例为阴囊内睾丸,其中4例行睾丸下降固定术9~27年后发生睾丸肿瘤。本研究1例患儿在13月龄发生双侧睾丸GCNIS,属于睾丸生殖细胞肿瘤的癌前病变,仅予观察,随访4年余暂未进展为生殖细胞肿瘤。PMDS患儿发生睾丸肿瘤的风险高,发生年龄可早至婴幼儿期,且睾丸下降固定术后仍可进展。
目前,对PMDS患儿MRs的处理存在争议。考虑到PMDS患儿MRs发生恶变率低,且男性输精管紧邻MRs或包埋于其内部,切除MRs过程中极易损伤输精管及相应血管,对生育功能造成不可逆损伤,因此大部分学者认为应当保留MRs或仅做部分切除[25,26]。然而,随着MRs发生恶变的相继报道,一部分学者认为应予以切除MRs[27]。
综上所述,PMDS发生睾丸肿瘤的风险远远高于其他隐睾患儿,睾丸下降不全患儿中应注意识别PMDS,并尽早行隐睾下降固定,并结合睾丸病理及临床激素变化长期严密监测。
所有作者均声明不存在利益冲突

























