
尿道下裂的全球发病率约为千分之二,是男性生殖系统中最常见的复杂先天性疾病之一。尿道下裂受遗传因素和环境因素共同作用。遗传是尿道下裂的主要病因,其遗传度约为57%~77%,但仅约30%的患儿获得了明确的遗传分子诊断。目前,已知数十个基因的遗传变异与尿道下裂风险相关,同时针对部分候选基因进行了小鼠模型研究,但尿道下裂的遗传病因仍不清楚。本研究就尿道下裂的遗传风险基因、动物模型、分子调控机制以及最新的技术进展进行总结和概述,以加深对尿道下裂发病机制的认识,从而为尿道下裂的预防和诊治提供新的参考方案。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
尿道下裂是一种常见的先天性泌尿生殖系统畸形,其全球发病率约为千分之二,近年来呈上升趋势[1],主要由孕8~14周的阴茎发育受干扰所导致[2]。尿道下裂表现为尿道开口异常,部分患儿伴阴茎或包皮异常,按照尿道开口位置可分为近端型(重度)、中端型(中度)和远端型(轻度)[3]。当前,手术是治疗尿道下裂的唯一方式,且近段型尿道下裂常需多次手术[2]。尿道下裂不仅影响患儿的排尿与生殖功能,也会给患儿及家属带来心理负担。
尿道下裂由遗传因素与环境因素共同导致(图1),其中遗传是主要病因,包括生殖结节发育相关的基因以及导致雄激素、雌激素紊乱的相关基因。尿道下裂的遗传度约为57%~77%[4,5]。此外,环境因素也是导致尿道下裂发生的重要因素,包括外源化学物质、避孕药、辅助生殖技术、妊娠期高血压及保胎药物等母体因素和低出生体重。在这些病因中,又以环境因素中的内分泌干扰物(endocrine disrupting chemicals, EDCs)和妊娠期高血压等母体因素以及遗传因素中参与生殖结节及男性化的多种基因及信号通路为研究重点[3,6]。目前,除了探究新的致病基因,构建动物模型也有助于研究致病因素在尿道下裂中的作用机制。至今为止,尿道下裂的具体分子调控机制仍不清楚。
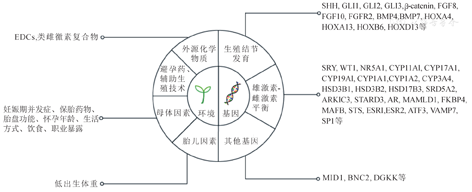
本研究将重点讨论与尿道下裂发病机制相关的遗传学因素,并对尿道下裂遗传因素相关的动物模型以及最新的技术进展进行总结和概述,以加深对尿道下裂发病机制的认识,从而为尿道下裂的预防和诊治提供新的参考方案。
人类男性外生殖器于孕5、6周至第8周之间的发育为激素非依赖性,从第8周受雄激素影响开始男性化,至第16、17周完成性别分化[7,8,9]。因而,外生殖器的发育不仅受多种生殖结节发育相关基因的影响,同时也与影响雄激素-雌激素平衡的基因有关。当前针对人类尿道下裂已开展大量的遗传学研究,以基于全基因组关联研究(genome-wide association study, GWAS)的单核苷酸多态性(single-nucleotide polymorphisms, SNPs)寻找为主,并基于GWAS发现了许多与尿道下裂风险相关的基因及信号通路。
参与生殖结节发育的基因及信号通路的异常与人类尿道下裂密切相关,目前已在SHH(Sonic HedgeHog)基因、GLI转录因子家族、成纤维细胞生长因子(fibroblast growth factor, FGF)信号通路、BMP基因以及同源异形盒基因(homeobox genes,HOX)中发现了尿道下裂相关易感位点或基因突变[10,11,12,13]。其中,SHH为HH(HedgeHog)家族成员,在Patched受体(Ptch)和Gli1-3的参与下进行信号转导[14],作用于生殖结节的生长、构型和细胞生存[15];FGF信号可维持细胞正常生存、分化和正常细胞黏附,调控早期生殖结节发育以及尿道成管过程[16,17];BMP基因在生殖结节发育时表达于间质及尿道上皮,通过调节细胞凋亡对外生殖器发育产生影响[18];HOX分子家族中的Hoxa13作用于泌尿生殖道终端的形态发生[19],并与同家族的Hoxd13共同在SHH的调节下通过调控泄殖腔形态发生参与生殖器结节发育[20]。
性腺发育相关基因的遗传变异是男性性发育异常(disorder of sex development, DSD)的重要原因,同时也常见于尿道下裂患儿。SRY基因突变是导致男性性腺发育不全的重要原因,同时SRY基因突变的嵌合体患儿可表现出尿道下裂及隐睾等外生殖器异常[21]。WT1突变不仅与Denys-Drash综合征以及Frasier综合征等具有性腺发育不良及外生殖器发育异常表现的疾病有关[22],同时也与孤立性尿道下裂有关[13]。NR5A1的致病性突变存在于46, XY DSD患儿以及尿道下裂患儿中[23,24]。
①雄激素合成及代谢相关基因
雄激素生物合成涉及多种酶,编码这些酶的基因产生的突变可能通过影响雄激素水平导致男性化减弱,从而表现出尿道下裂表型。目前,CYP11A1, HSD3B1, HSD3B2, HSD17B3, SRD5A2, AKR1C3等基因中的突变已在尿道下裂患儿中被发现[25,26,27,28];CYP17A1, HSD17B3, HSD3B1, SRD5A2以及AKR1C3等基因存在与尿道下裂相关的易感位点[28,29,30]。然而,这些基因变异在尿道下裂中所起的作用仍需观察,因为有研究认为部分发现于尿道下裂患儿的SRD5A2突变可能与尿道下裂无关联[13]。此外,近期的研究亦表明雄激素代谢相关基因CYP19A1, CYP3A4, HSD17B14, HSD3B7, HSD17B7及CYP11A1的异常表达与尿道下裂风险相关[26]。调控睾酮分泌的基因也可能与尿道下裂有关,如STARD3和MAMLD1(CXorf6)均存在尿道下裂风险位点[13, 30],并且在尿道下裂患儿中还发现了位于MAMLD1基因的突变[27]。
②雄激素受体
雄激素受体(androgen receptor, AR)在外生殖器男性化中起关键作用,其遗传变异可能影响雄激素的正常作用,导致尿道下裂等男性化不足表型。目前已在各类尿道下裂患者中发现AR突变[27],并且AR基因中SNP位点rs5919436[31]及CAG重复长度变异[13]也可能影响尿道下裂患病风险。此外,与AR发生相互作用的基因也可能通过网络影响尿道下裂发生[32]。
①雌激素合成及代谢相关基因
外源雌激素暴露是尿道下裂的危险因素之一[3]。雌激素合成及代谢相关基因同样可影响雌激素水平,在这些基因中,雌激素合成相关的CYP19A1基因的突变可能与男患儿的尿道下裂有关,作用于雌激素代谢的CYP1A1, CYP1A2, CYP3A4,以及参与具有生物活性雌激素合成的STS基因中存在尿道下裂易感位点[13, 29]。
②雌激素受体和相关基因
雌激素受体(estrogen receptor, ER)介导雌激素信号的传导,其相关基因的遗传变异也是导致尿道下裂的发病原因之一。雌激素受体有ESR1和ESR2两种亚型,其中在ESR1基因中已发现多个尿道下裂易感位点及风险单倍型域[33,34],ESR2中除存在尿道下裂风险位点,其CA重复序列增加也会增加尿道下裂的风险[35,36]。另有研究提示,在部分尿道下裂患儿中发现的ER变异可降低尿道下裂风险或与尿道下裂不存在风险相关性,因此ER在尿道下裂发病机制中的作用仍需深入研究[13, 33]。
多种雌激素敏感基与尿道下裂密切相关。CYR61, ATF3, CTGF, GADD45β, ZEB1等雌激素敏感基因在尿道下裂患儿包皮中表达增加[37,38,39],其中ZEB1在重度尿道下裂患儿包皮中上调更为明显[37],ATF3基因中的SNP位点rs11119982则在宫内细胞因子暴露的情况下使胎儿尿道下裂患病风险升高[40]。与ER发生相互作用的基因也可能与尿道下裂等泌尿系统畸形相关。VAMP7可调控雌激素受体,包含此基因的一段X染色体区域的重复可导致先天性泌尿生殖道男性化异常(其中包括尿道下裂表型)[41]。SP1除与AR发生相互作用,也可与ER和ATF3相互作用,参与雌激素信号调控[32]。
除了上述基因,还有其他基因也可能参与尿道下裂发病机制。有研究表明,MID1,BNC2,二酰基甘油激酶κ(DGKK),IRX5,IRX6和EYA1等基因上存在尿道下裂风险位点[11, 27, 42],这些新发现的尿道下裂候选基因在尿道下裂发病机制中所起的作用有待进一步研究。此外,其他在尿道下裂患儿中差异表达的基因也可能与尿道下裂有潜在的联系,它们与凋亡、代谢、蛋白结合、受体活性、信号传导、转录、翻译和载体活性相关[39]。
为了对人类尿道下裂的发病机制进行探究,研究者们构建了尿道下裂的动物模型。尿道下裂的动物模型以小鼠模型为多见[43]。目前,为了研究尿道下裂相关的遗传学因素,研究者们构建了多种人类尿道下裂相关基因的敲除或过表达的小鼠模型(表1)。多种基因的小鼠敲除模型表现出与人类相似的尿道下裂表型,包括SHH, GLI2, GLI3, FGF10, AR, CYP19A1, ESR1, BNC2等基因。此外,BMP4, BMP7, SRY, WT1, CYP11A1, EMX2等基因的小鼠模型则表现出较尿道下裂更严重的泌尿生殖系缺陷。目前为止,SRD5A2, MAMLD1, ATF3, GLI3, CTGF等人类尿道下裂相关基因仍未能在小鼠中与尿道下裂成功建立联系,并且β-catenin, FKBP4, HOXA13, Ephrin-B2, EphB2, MAFB, LAMA5, SPRY1/SPRY2, VAMP7等基因仅在小鼠模型而非人类中与尿道下裂相关,这种不一致性可能是由人类和小鼠外生殖器发育机制的差异所导致。(表1)
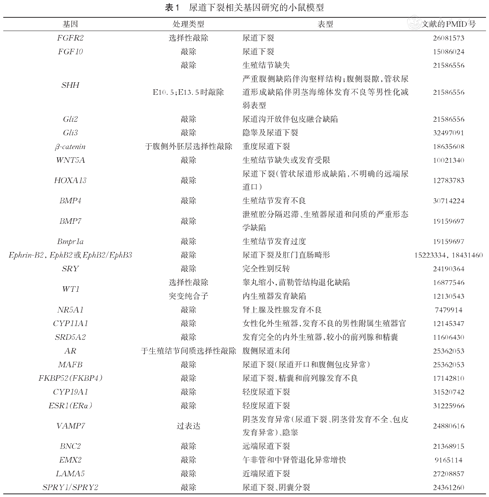
尿道下裂相关基因研究的小鼠模型
尿道下裂相关基因研究的小鼠模型
| 基因 | 处理类型 | 表型 | 文献的PMID号 |
|---|---|---|---|
| FGFR2 | 选择性敲除 | 尿道下裂 | 26081573 |
| FGF10 | 敲除 | 尿道下裂 | 15086024 |
| SHH | 敲除 | 生殖结节缺失 | 21586556 |
| E10.5;E13.5时敲除 | 严重腹侧缺陷伴沟壑样结构;腹侧裂隙,管状尿道形成缺陷伴阴茎海绵体发育不良等男性化减弱表型 | 21586556 | |
| Gli2 | 敲除 | 尿道沟开放伴包皮融合缺陷 | 21586556 |
| Gli3 | 敲除 | 隐睾及尿道下裂 | 32497091 |
| β-catenin | 于腹侧外胚层选择性敲除 | 重度尿道下裂 | 18635608 |
| WNT5A | 敲除 | 生殖结节缺失或发育受限 | 10021340 |
| HOXA13 | 敲除 | 尿道下裂(管状尿道形成缺陷,不明确的远端尿道口) | 12783783 |
| BMP4 | 敲除 | 生殖结节发育不良 | 30714224 |
| BMP7 | 敲除 | 泄殖腔分隔迟滞、生殖器尿道和间质的严重形态学缺陷 | 19159697 |
| Bmpr1a | 敲除 | 生殖结节发育过度 | 19159697 |
| Ephrin-B2,EphB2或EphB2/EphB3 | 敲除 | 尿道下裂及肛门直肠畸形 | 15223334,18431460 |
| SRY | 敲除 | 完全性别反转 | 24190364 |
| WT1 | 选择性敲除 | 睾丸缩小,苗勒管结构退化缺陷 | 16877546 |
| 突变纯合子 | 内生殖器发育缺陷 | 12130543 | |
| NR5A1 | 敲除 | 肾上腺及性腺发育不良 | 7479914 |
| CYP11A1 | 敲除 | 女性化外生殖器,发育不良的男性附属生殖器官 | 12145347 |
| SRD5A2 | 敲除 | 发育完全的内外生殖器,较小的前列腺和精囊 | 11606430 |
| AR | 于生殖结节间质选择性敲除 | 腹侧尿道未闭 | 25362053 |
| MAFB | 敲除 | 尿道下裂(尿道开口和腹侧包皮异常) | 25362053 |
| FKBP52(FKBP4) | 敲除 | 尿道下裂,精囊和前列腺发育不良 | 17142810 |
| CYP19A1 | 敲除 | 轻度尿道下裂 | 31520742 |
| ESR1(ERα) | 敲除 | 轻度尿道下裂 | 31225966 |
| VAMP7 | 过表达 | 阴茎发育异常(尿道下裂、阴茎骨发育不全、包皮发育异常)、隐睾 | 24880616 |
| BNC2 | 敲除 | 远端尿道下裂 | 21368915 |
| EMX2 | 敲除 | 午非管和中肾管退化异常增快 | 9165114 |
| LAMA5 | 敲除 | 近端尿道下裂 | 27208857 |
| SPRY1/SPRY2 | 敲除 | 尿道下裂、阴囊分裂 | 24361260 |
在构建尿道下裂动物模型时,应重视人类与动物模型在生殖器发育上存在的差异。在小鼠和大鼠阴茎中,近端尿道由尿道板直接成管形成,而远端尿道涉及腹侧融合事件,这与人类的尿道发育规律相反[44,45]。人类尿道下裂主要与尿道的融合事件相关,动物模型的"尿道下裂"表型要对应上融合事件相关的外生殖器发育异常,如鼠类尿道口及男性泌尿生殖道交配突起(male urogenital mating protuberance,MUMP)等结构的发育缺陷;并且,由于小鼠等动物的尿道发育持续至出生后,一部分在胚胎期或出生时表现出尿道畸形的动物模型最终发育为正常表型,因此在尿道发育完成后保持尿道下裂表型的动物模型可能更具说服力[43]。
对人类尿道下裂遗传及动物模型的研究表明尿道下裂是受多基因调控的复杂疾病,且常伴发其他疾病和表型。目前对人类以及动物模型的研究表明,外生殖器男性化依赖雄激素信号和雌激素信号的共同作用,雄激素-雌激素的异常调控将导致尿道下裂等男性化减弱表现。雄激素(睾酮或二氢睾酮)通过与AR结合,激活雄激素敏感基因,实现雄激素信号传导。AR在融合中的尿道褶间质中表达,提示雄激素信号作用于尿道形成中的融合事件[46]。
同雄激素信号相类似,雌激素信号通过雌激素与ER的结合进行传导。雌激素信号对阴茎发育的影响具有两面性。一方面,雌激素信号可能参与阴茎的正常发育;另一方面,过度的雌激素信号会破坏雄激素-雌激素平衡,阻碍外生殖器男性化的正常进行[8]。本团队前期对尿道下裂的网络互作和遗传研究亦表明尿道下裂中存在以AR为核心的调控网络,与AR和ER存在互作基因的遗传变异可以通过该网络影响尿道下裂的发生[32]。此外,尿道下裂的表型与包括AR在内的特定的转录调控模式有关[47]。理解尿道发育过程中以AR为核心的相关基因的遗传变异及特征调控网络将进一步夯实尿道下裂的遗传分子调控基础。
尿道下裂呈现多基因遗传模式。以往针对尿道下裂的研究主要以GWAS技术研究为主,但仅解释了尿道下裂约9%的遗传度[11],而二代测序(next-generation sequencing, NGS)技术则可以对尿道下裂中的罕见突变进行高通量检测以弥补GWAS所带来的遗传度缺失。近期,Ea等[48]利用二代测序技术在大队列尿道下裂中检测致病突变,但由于该项研究仅限于336个候选基因,且主要关注有害的错义突变,这导致很多功能丧失型突变未被统计在内,同时他们的研究也造成了遗传统计学证据的缺失以及遗传度缺失。我们在此基础上进一步提出利用"大效应的稀有变异"新思路可以补充现有方法研究尿道下裂造成的遗传度缺失[49],通过比较不同人群的最小等位基因频率(minor allele frequency, MAF)筛选进化上保守的,同时对蛋白质功能可能会产生重要影响的致病突变,这将为进一步寻找尿道下裂的致病机制提供理论基础。
基因表观遗传的改变也是导致人类尿道下裂的一个不可忽视的因素。Vottero等[50]首先提出,尿道下裂患儿包皮中AR的甲基化水平高于正常对照。此后,研究者们为进一步探究其他基因的甲基化与尿道下裂之间的关系,进行了相应的表观基因组关联研究(epigenome-wide association studies, EWAS)。近期,Richard等[51]通过EWAS发现了25个新的尿道下裂相关甲基化位点,涉及胚层分化、β-catenin信号通路、雄激素以及生殖特征相关的多个基因。这种表观遗传的改变可能是环境因素诱导尿道下裂的一种机制,如己烯雌酚对人类和小鼠尿道下裂风险的影响可持续至第二代[3],这种效应可能与己烯雌酚等环境雌激素通过诱导非基因组ER信号传导以激活PI3K/AKT而造成的组蛋白甲基化改变有关[52]。
目前人们对尿道下裂遗传因素的研究已经取得了一定进展,然而由于尿道下裂的成因较为复杂,候选基因在尿道下裂中的具体作用机制仍不明确。随着遗传学技术的迅速发展,新的尿道下裂致病位点也在不断被发掘,这些新发现有助于完善人们对尿道下裂相关风险基因致病机制的认识。同时,尿道下裂动物模型的发展有力地促进了尿道下裂相关分子机制的研究,其中基因编辑技术的快速发展也推进了对风险基因在尿道下裂过程中分子作用机制的理解进程,人们对尿道下裂遗传因素的理解将更为深刻,有利于尿道下裂的预防及诊治。
所有作者均声明不存在利益冲突

























