
腓骨肌萎缩症是最常见的一组遗传性周围神经病,临床特点包括对称性远端为主的肌无力伴萎缩、感觉减退以及弓形足、脊柱侧凸等骨骼畸形。临床上根据神经电生理、病理学和遗传学特点可分为多种亚型,基因检测手段有助于明确其致病基因。康复治疗、外科矫形手术治疗和改善症状的药物治疗有助于缓解疾病症状,减轻骨骼畸形。部分针对病因和发病机制的特异性治疗药物已进入临床试验阶段,其疗效和安全性有待进一步明确。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2024年共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。本年度继教答题得学分活动将于10月20日结束。
腓骨肌萎缩症(Charcot-Marie-Tooth diseases,CMT)又称为遗传性运动感觉神经病(hereditary motor and sensory neuropathy),是一组最常见的周围神经单基因遗传病,由法国的Charcot和Marie以及英国的Tooth在1886年首先报道并系统描述[1]。CMT的患病率约为40/100 000,临床特点包括:儿童或青少年起病,慢性进行性的对称性肌无力及肌萎缩、远端型感觉障碍、腱反射减弱或消失、弓形足等骨骼畸形[1]。CMT具有显著的遗传异质性和临床异质性,相同的基因突变可导致不同的临床表型,目前已有大约100个不同的CMT致病基因相继被克隆并报道[2]。
临床上根据神经电生理和神经病理学的特点,将CMT分为以下3种类型:(1)脱髓鞘型CMT:以显著的神经传导速度(nerve conduction velocity,NCV)减慢为特点,通常上肢运动NCV<38 m/s,神经活组织检查(活检)病理结果提示显著的周围神经髓鞘异常。(2)轴索型CMT:周围神经NCV轻度减慢或接近正常,通常上肢运动NCV>38 m/s,神经活检病理结果提示慢性轴索变性及再生。(3)中间型CMT:上肢运动NCV在25~45 m/s。CMT的遗传方式多数呈常染色体显性遗传,也可呈常染色体隐性遗传及X连锁显性或隐性遗传。结合遗传方式、临床表现、电生理和病理特点,可将CMT进一步分为以下几种亚型[1](表1)。
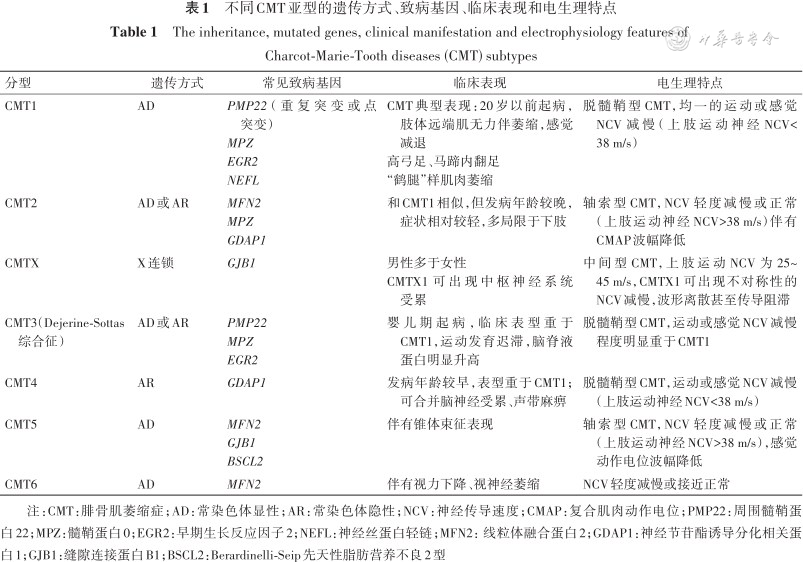
不同CMT亚型的遗传方式、致病基因、临床表现和电生理特点
The inheritance, mutated genes, clinical manifestation and electrophysiology features of Charcot-Marie-Tooth diseases (CMT) subtypes
不同CMT亚型的遗传方式、致病基因、临床表现和电生理特点
The inheritance, mutated genes, clinical manifestation and electrophysiology features of Charcot-Marie-Tooth diseases (CMT) subtypes
| 分型 | 遗传方式 | 常见致病基因 | 临床表现 | 电生理特点 |
|---|---|---|---|---|
| CMT1 | AD | PMP22(重复突变或点突变) MPZ EGR2 NEFL | CMT典型表现:20岁以前起病,肢体远端肌无力伴萎缩,感觉减退 高弓足、马蹄内翻足 “鹤腿”样肌肉萎缩 | 脱髓鞘型CMT,均一的运动或感觉NCV减慢(上肢运动神经NCV<38 m/s) |
| CMT2 | AD或AR | MFN2 MPZ GDAP1 | 和CMT1相似,但发病年龄较晚,症状相对较轻,多局限于下肢 | 轴索型CMT,NCV轻度减慢或正常(上肢运动神经NCV>38 m/s)伴有CMAP波幅降低 |
| CMTX | X连锁 | GJB1 | 男性多于女性 CMTX1可出现中枢神经系统受累 | 中间型CMT,上肢运动NCV为25~45 m/s,CMTX1可出现不对称性的NCV减慢,波形离散甚至传导阻滞 |
| CMT3(Dejerine-Sottas综合征) | AD或AR | PMP22 MPZ EGR2 | 婴儿期起病,临床表型重于CMT1,运动发育迟滞,脑脊液蛋白明显升高 | 脱髓鞘型CMT,运动或感觉NCV减慢程度明显重于CMT1 |
| CMT4 | AR | GDAP1 | 发病年龄较早,表型重于CMT1;可合并脑神经受累、声带麻痹 | 脱髓鞘型CMT,运动或感觉NCV减慢(上肢运动神经NCV<38 m/s) |
| CMT5 | AD | MFN2 GJB1 BSCL2 | 伴有锥体束征表现 | 轴索型CMT,NCV轻度减慢或正常(上肢运动神经NCV>38 m/s),感觉动作电位波幅降低 |
| CMT6 | AD | MFN2 | 伴有视力下降、视神经萎缩 | NCV轻度减慢或接近正常 |
注:CMT:腓骨肌萎缩症;AD:常染色体显性;AR:常染色体隐性;NCV:神经传导速度;CMAP:复合肌肉动作电位;PMP22:周围髓鞘蛋白22;MPZ:髓鞘蛋白0;EGR2:早期生长反应因子2;NEFL:神经丝蛋白轻链;MFN2:线粒体融合蛋白2;GDAP1:神经节苷酯诱导分化相关蛋白1;GJB1:缝隙连接蛋白B1;BSCL2:Berardinelli-Seip先天性脂肪营养不良2型
CMT1为常染色体显性遗传的脱髓鞘型CMT,是CMT中最常见的一型。其中,CMT1A是最常见的CMT亚型,占所有CMT的40%~50%,多数与染色体17p11.2-12上的包含周围髓鞘蛋白22(peripheral myelin protein 22,PMP22)基因1.4 Mb片段的重复突变相关[3]。在少数情况下,CMT1A由PMP22基因点突变所导致。CMT1B与髓鞘蛋白0(myelin protein zero,MPZ)基因点突变相关,约占CMT1总数的3%~5%。其他常见的致病基因还包括早期生长反应因子2(early-growth-response 2,EGR2)、神经丝蛋白轻链(neurofilament light chain,NEFL)基因等。
CMT2为轴索型CMT,可呈常染色体显性或隐性遗传,具有高度的遗传异质性。常染色体显性遗传的CMT2最常见的致病基因为线粒体融合蛋白2(mitofusin 2,MFN2),占CMT2病例总数的20%;其次为MPZ基因,约占CMT2病例总数的5%。常染色体隐性遗传的CMT2最常见的致病基因为神经节苷酯诱导分化相关蛋白1(ganglioside-induced differentiation-associated protein 1,GDAP1)。
X连锁隐性遗传的CMTX1是第二常见的CMT亚型,约占所有CMT的10%,男性患者较女性更为常见,绝大多数与缝隙连接蛋白B1(gap junction B1,GJB1)基因突变相关。电生理表现符合中间型CMT的特点。
CMT3又称为Dejerine-Sottas综合征,呈常染色体显性或隐性遗传,发病年龄较早,是临床表型最严重的脱髓鞘型CMT,致病基因包括PMP22、MPZ、EGR2基因。
CMT4为常染色体隐性遗传的脱髓鞘型CMT,最常见的致病基因为GDAP1。
CMT5呈常染色体显性遗传,通常合并锥体束受累的表现,电生理表现符合轴索型CMT特点。致病基因包括MFN2、GJB1、Berardinelli-Seip先天性脂肪营养不良2型(Berardinelli-Seip congenital lipodystrophy type 2,BSCL2)。
CMT6呈常染色体显性遗传,可出现视神经萎缩的表现,NCV轻度减慢或接近正常。致病基因为MFN2。
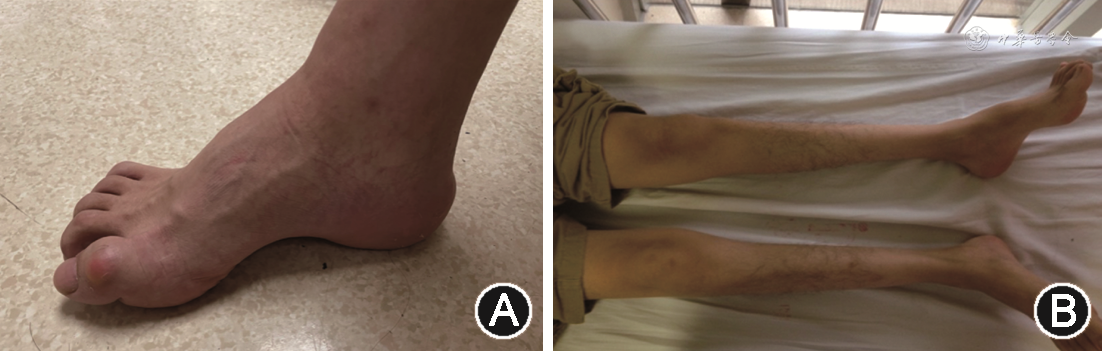
CMT多于20岁之前起病,少数情况下可在婴儿期发病,主要临床表现为肢体远端为主的肌肉无力伴萎缩,感觉减退,症状向近端逐渐缓慢进展。典型的CMT运动症状多开始于足部,表现为高弓足(图1A)或扁平足、锤状趾,足背屈无力导致足下垂,严重时形成“马蹄内翻足”,患者可出现跑步困难、易绊倒、行走呈跨阈步态等表现。随着疾病的进展,大腿下1/3和小腿出现明显的肌肉萎缩,形似“鹤腿”(图1B)、“倒香槟酒瓶”样改变。这个时期也可出现前臂和手部肌肉无力、萎缩,手内在肌萎缩形似“爪形手”。感觉症状由肢体远端向近端逐渐发展,多累及足部和手部,表现为疼痛或感觉异常,体检可见痛触觉和振动觉减退,严重的本体感觉减退还可出现感觉性共济失调。除了足部畸形,患者还可出现脊柱侧弯等骨骼畸形。
不同亚型的CMT临床表现有所差异。CMT1型患者多表现为典型的CMT症状体征。CMT2型的临床症状体征与CMT1型相似,但发病年龄较晚,症状相对较轻,多局限于下肢,部分患者可伴有听力丧失、声带及膈肌麻痹、视神经萎缩、脑白质异常等表现。Dejerine-Sottas综合征的患者多于婴儿期起病,出现运动发育迟滞,严重的NCV减慢,脑脊液蛋白升高等表现[4]。CMTX1型男性和女性患者的临床表现差异较大,女性患者通常起病较晚,病情严重程度较男性患者轻,电生理表现上,运动神经NCV的减慢程度和轴索损害程度相对较轻[5]。由于女性CMTX1患者临床表现不典型,临床上容易与一些获得性周围神经疾病相混淆。新近的一项研究发现,女性CMTX1型患者无症状的比例显著高于男性患者,有症状的女性患者起病较早(<19岁)者临床症状较严重;中年起病的女性患者(>48岁)中近半数患者(45%)症状较轻微。有4例女性患者在基因明确诊断为CMTX1型前,还被误诊为类似慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)而接受静脉免疫球蛋白治疗[6]。还有研究发现CMTX1患者可多次出现卒中样发作的偏侧肢体无力症状,而且MRI的异常信号呈现空间波动性,在随访5个月的时间内发现MRI的异常信号可由双侧脑白质转移至胼胝体,最后消失[7]。CMT4型患者发病年龄较早,症状体征较重,可合并脑神经受累、声带麻痹等。CMT5型患者伴有锥体束征表现。CMT6型患者常伴有视力下降、视神经萎缩[8]。
随着基因检测手段的发展,越来越多潜在的CMT致病基因及新的突变位点被发现,CMT的基因型和临床表型谱也在不断丰富和扩展。在最新发表的研究中,我国吴志英团队报道了COQ7基因新发复合杂合突变导致辅酶Q10合成障碍从而引起轴索型CMT的病例,患者的临床表现近似CMT2型[9]。在此之前已有研究报道COQ7基因其他突变位点可导致远端遗传性运动神经病(distal hereditary motor neuronopathy,dHMN)、遗传性痉挛性截瘫等表现[10, 11]。
神经电生理检查尤其是NCV检测能够协助明确周围神经脱髓鞘或轴索损害,有助于CMT的分型。均一的NCV减慢(上肢正中神经或尺神经运动传导速度<38 m/s)提示脱髓鞘型CMT(CMT1和CMT4)。NCV轻度减慢或正常(上肢正中神经或尺神经运动传导速度>38 m/s)伴有复合肌肉动作电位或感觉动作电位波幅降低提示典型的CMT2。当NCV提示中间型(上肢运动NCV为25~45 m/s),对于男性患者需注意CMTX可能[12]。其次,不论男性或女性患者还需注意显性遗传的中间型CMT可能(相比CMTX更为罕见)。肌电图检查通常表现为时程长、波幅高的运动单位电位,并且募集相减少,纤颤电位较少见,提示慢性失神经支配和神经再生现象。
NCV检查还有助于鉴别CMT和其他获得性多发性周围神经病。在CMT1中,NCV减慢十分明显且基本对称,通常没有运动神经的传导阻滞或动作电位波形离散;而获得性脱髓鞘性周围神经病如CIDP的NCV减慢并不十分明显,且通常不对称,可见运动神经传导阻滞或动作电位波形离散。但需要注意的是,CMTX1型患者可能出现不对称性的NCV减慢,以及波形离散甚至传导阻滞的情况。在更为罕见的情况下,MPZ基因突变所致的CMT1B也可出现传导阻滞[13]。电生理检查还有助于鉴别CMT及其他神经肌肉疾病例如远端型肌病/肌营养不良、dHMN等。
基因检测是明确诊断CMT十分重要的手段。随着二代测序技术包括疾病特异性基因检测、全外显子测序、全基因组测序、高通量转录组测序等在临床应用的普及,越来越多的CMT致病基因和突变位点被发现。PMP22、MPZ、GJB1、MFN2和GDAP1是其中最重要的致病基因。在常染色体显性遗传CMT1和散发CMT1患者中,应首先利用多重连接探针扩增技术(multiplex ligation dependent probe amplification)进行PMP22基因大片段重复突变检测。如果检测结果为阴性且家系内无男传男现象,应考虑CMTX可能并进行GJB1基因的突变分析。若除外CMTX1可能,还应进行PMP22和MPZ的点突变检测。在考虑为CMT2诊断的患者中,应首先进行MFN2和MPZ基因突变检测。对家系内无男传男现象的病例,特别是女性CMT2病例,应考虑CMTX可能并进行GJB1基因突变分析。对于中间型NCV的患者,应首先进行GJB1、MPZ、NFL、GDAP1等基因的突变分析。在常染色体隐性遗传的CMT患者中,不论是脱髓鞘型(CMT4)和轴索型CMT2,均应首先进行GDAP1基因检测[14]。临床拟诊CMT的家系,若基因检测结果为阴性,应考虑是否可能为其他类型的遗传性周围神经病,如dHMN、遗传性感觉自主神经病(hereditary sensory and autonomic neuropathy)等,可进一步进行相关基因检测;或者可能存在未报道的新的CMT致病基因,应进行全外显子测序或全基因组测序,寻找可能的新的致病基因突变并进行功能验证[15, 16]。
随着基因检测技术的普及,神经活检病理检查在多数CMT病例的诊断中已非必须。然而,对于一些诊断困难的散发性病例或者基因检测阴性的家族性病例,周围神经活检病理结果可能会提供诊断上的帮助。光镜下观察周围神经的甲苯胺蓝染色可见有髓纤维大小不一,电镜下可见薄鞘纤维和裸轴索纤维,施万细胞增生形成“洋葱头”样结构提示CMT1。有髓和无髓纤维的减少、再生簇生成提示CMT2。CMTX可表现为轴索丢失和部分脱髓鞘改变并存,“洋葱头”样结构少见。某些特殊的髓鞘病理改变可能与特定的基因突变相关。例如,髓鞘松解与髓鞘肿胀可见于MPZ基因突变的患者;大量的髓鞘向外折叠是肌管微素相关蛋白2(myotubularin-related protein 2,MTMR2)基因、FGD1相关F肌动蛋白结合蛋白(FGD1-related F-actin binding protein,FGD4)基因突变所致的CMT4患者的典型病理改变;巨轴索可见于NEFL基因突变的病例[17]。
1.确定临床表型:大部分CMT亚型的临床表现符合典型的CMT症状体征,尤其是PMP22基因重复突变所致的CMT1A。应重视病史的询问和体格检查,许多临床特异性的表现对于后续基因检测策略具有重要的指向作用,包括起病年龄、病情严重程度以及某些特殊体征(如脑神经受累、声带麻痹、瞳孔异常、青光眼、视神经萎缩、锥体束受累、上肢受累为主、感觉异常为主等)。
2.确定遗传方式:根据病史采集尤其是家族史询问明确遗传方式为常染色体显性遗传、常染色体隐性遗传或X连锁遗传。常染色体显性遗传是CMT最常见的遗传方式,见于CMT1和大部分CMT2病例;CMTX1为X连锁显性遗传,家系内无男传男的表现,男性患者较女性患者症状严重;常染色体隐性遗传方式见于CMT4和少数CMT2病例。散发性CMT病例也不少见。由于CMT的临床表型异质性较大,同一个家系内的其他成员可能症状轻微甚至没有不适主诉,此时需要对先证者的其他家系成员进行详细的体格检查,必要时可进行NCV检测,有助于发现临床下的周围神经受累,为阳性家族史提供证据。
3.神经电生理检查:根据NCV的特点区分脱髓鞘型、轴索型或中间型。具体内容见辅助检查中神经电生理检查部分内容。
4.根据临床表现、遗传情况和电生理检测结果制订基因检测策略。具体内容见辅助检查中基因检测部分内容。
5.对于诊断困难的病例可选择性进行周围神经活检病理检测协助诊断。
本病的诊断依据如下:(1)儿童或青少年起病,可有阳性家族史。(2)缓慢进展的对称性肢体远端肌无力伴肌萎缩,典型表现为足部、小腿和大腿下1/3肌肉萎缩,可出现弓形足、锤状趾、脊柱侧弯等骨骼畸形,伴有感觉障碍,出现肢体远端对称性痛触觉、振动觉减退。体检腱反射对称性减弱或消失。(3)神经电生理检查结果提示运动和感觉神经脱髓鞘损害或轴索损害表现。(4)神经活检病理结果提示脱髓鞘、髓鞘再生或轴索变性特点。(5)基因检测可协助明确CMT的临床诊断和分型。
CMT需要与获得性多发性周围神经病、一些以周围神经受累为主的遗传性疾病或一些累及周围神经的其他遗传性疾病相鉴别。
CIDP是一类由免疫介导的运动感觉周围神经病,好发于20~60岁,男女发病比例相近,病程慢性进展或呈复发缓解,临床表现为不同程度的对称性肢体无力,少数为非对称性,近端和远端均可累及,四肢腱反射减低或消失,伴有深、浅感觉异常。脑脊液蛋白明显升高,细胞数正常,呈现蛋白-细胞分离现象。神经电生理检查表现为周围NCV减慢、传导阻滞及异常波形离散。病理显示有髓纤维多灶性脱髓鞘、神经内膜水肿、炎性细胞浸润等特点,大部分病例对激素治疗有效[18]。
FAP多于20~50岁逐渐起病,1~2年内进展迅速,呈常染色体显性遗传。临床表现为双下肢感觉异常及疼痛,伴肌无力,感觉和自主神经损害较突出,常伴有内脏损害。临床上分为4型,各亚型之间有交叉,各型均为男性多于女性。心脏彩超和心电图检查提示心肌受累。脑脊液检查可见蛋白增高。肌电图检查结果提示周围神经神经源性损害。神经活检可发现淀粉样物质沉积。基因检测发现转甲状腺素蛋(transthyretin,TTR)、载脂蛋白AI(apolipoprotein AI,APOAI)、凝溶胶蛋白(gelsolin,GSN)等基因突变可确诊[19]。
dHMN又称为远端型脊肌萎缩症,发病年龄跨度大,婴幼儿、儿童、青少年及成年人均可发病,主要表现为四肢远端肌无力和萎缩,不伴有感觉减退。神经电生理检查是该病与CMT鉴别诊断的关键。dHMN患者运动和感觉NCV检查正常,肌电图提示前角细胞损害特征。临床上可分为多个亚型,各个分型之间表现各有不同。dHMNⅡ型是常染色体显性遗传的dHMN经典型,由热休克蛋白27(heat shock 27-kDa protein 1,HSP27)和热休克蛋白22(heat shock 22-kDa protein 8,HSP22)基因突变导致,又称为CMT2F及CMT2L。dHMNⅥ型较罕见,但表型较重,由免疫球蛋白结合蛋白(immunoglobulin mu binding protein 2,IGHMBP2)基因突变导致,呈常染色体隐性遗传,表现为婴幼儿期呼吸肌及远端肌受累(又称脊髓型肌萎缩伴呼吸衰竭Ⅰ型)。dHMNⅦ型由动力肌动蛋白(dynactin,DCTN1)基因突变导致,主要表现为进行性的面部、四肢肌无力、萎缩伴声带麻痹[20]。
HNPP通常为青少年起病(平均19~26岁),无性别差异,多由PMP22基因大片段缺失所导致,呈常染色体显性遗传。主要表现为复发性、无痛性多神经病,牵拉或受压后反复出现肢体麻木、无力,通常为非对称性,经数天至数周缓解。肌电图提示弥漫性NCV减慢。神经活检病理可见周围神经节段性脱髓鞘和局灶性髓鞘增厚,形似“腊肠样”结构。基因检测有助于明确诊断[21]。
遗传性共济失调性多发性神经病又称Refsum病,植烷酸贮积症,主要由于植烷酸-CoA-羟化酶缺陷,体内大量植烷酸贮积致病,呈常染色体隐性遗传。约1/3在10岁前发病,半数在10~30岁发病。逐渐起病,缓慢进展,临床表现为夜盲与视网膜色素变性、多发性神经病、小脑共济失调3大主征。血植烷酸含量增高,脑脊液蛋白异常增高,基因分析有助诊断[22]。
远端型肌病/肌营养不良是一组以进行性四肢远端向近端逐渐发展的肌无力、萎缩为临床特点等遗传性骨骼肌疾病,肌电图呈肌源性损害,运动和感觉NCV正常。肌肉活检病理提示肌源性损害[23]。
目前尚无有效逆转CMT病程的治疗方法,支持性治疗包括康复治疗、外科矫形手术治疗和缓解症状为主的药物治疗等,同时需要多学科团队相互协作[24]。已有研究针对不同CMT亚型的发病机制和治疗靶点研发了相应的特异性治疗药物,但绝大多数处于药物临床试验阶段,尚未进入临床治疗[25]。
多种康复治疗手段已被用于CMT的治疗。有研究结果表明,轻到中等强度的运动训练能有助于CMT患者改善下肢肌力及行走能力。适当的有氧训练能够改善患者的体能,提高有氧运动功能。下肢的被动拉伸训练有助于预防和改善跟腱挛缩。适当穿戴辅助器具如矫形器能够改善患者的姿势和平衡功能。个体化的踝-足矫形器有助于改善足下垂,辅助患者行走并缓解足弓疼痛[26]。
多数患者在儿童和青少年时期足部的内翻畸形是柔软可恢复的,此时应首先选择非手术治疗。随着病情的发展,足内翻逐步进展加重为固定畸形,无法穿戴矫形器具,此时需要采取外科手术治疗[27]。手术治疗的原则是纠正足部畸形,重建和平衡足踝肌力。手术方案包括肌力平衡手术(各种类型的肌腱转移术)、软组织松解与矫形(足底筋膜切开术)、跟骨截骨矫形、腓肠肌复合体处理、踇趾矫形或其他足趾矫形手术[28]。
疼痛是CMT患者常见的症状,可见于23%~85%的患者,可能与骨骼畸形、长期姿势异常或肌疲劳相关[29]。疼痛的治疗除了物理康复治疗、穿戴矫形器、外科矫形手术外,还可以使用非甾体抗炎药例如对乙酰氨基酚等缓解疼痛。对于明确的神经性疼痛的患者,可以使用一线或二线缓解神经病理性疼痛的药物例如三环类抗抑郁药、选择性五羟色胺再摄取抑制剂(selective serotonin reuptake inhibitors)类、抗惊厥药(如普瑞巴林、加巴喷丁、卡马西平等),尽量避免使用阿片类药物[30]。
CMT患者也常常出现疲劳感,可能与肌力下降、心肺功能降低有关,部分患者可能与阻塞性睡眠呼吸暂停综合征有关。有研究结果表明莫达非尼治疗能缓解CMT患者的疲劳感,但需注意相关的不良反应[31]。另外,应注意避免使用可能加重周围神经损害的药物,例如长春新碱、铂类、紫杉醇衍生物、硼替佐米、呋喃妥因、氨苯砜、来氟米特、甲硝唑、司他夫定、他克莫司等。
目前针对CMT发病机制中相关靶点的治疗研究多集中在PMP22基因突变所致的CMT1A[32]。维生素C在体外细胞实验研究中被证实能够促进髓鞘形成。动物实验研究表明,高剂量维生素C治疗能够改善CMT1A模型鼠的运动功能。然而,在临床试验中,不同剂量的维生素C(1~4 g/d)治疗并不能有效改善CMT1A患者的临床表型。维生素C治疗CMT1A是否有效还需后续更多临床试验进一步研究。动物实验研究结果表明黄体酮及其衍生物可加重CMT1A大鼠的神经功能缺损症状和神经病理损害。黄体酮拮抗剂——奥司那酮(onapristone)被证实能够降低CMT1A大鼠周围神经PMP22蛋白的表达并改善疾病表型[33]。然而,由于难以耐受的毒副作用,奥司那酮无法应用于CMT1A患者的临床治疗。ulapristal是奥司那酮的生物等效剂,具有较好的安全性。在法国开展的ulapristal治疗CMT1A的Ⅱ期临床研究正在进行中。神经营养因子3(neurotrophin-3,NT3)能够促进轴突生长,在一项随机、双盲、安慰剂对照试验中,8例CMT1A患者接受了6个月的皮内NT3注射治疗,NT3似乎能够改善患者的感觉神经受累程度,增加腓肠神经小直径有髓纤维的数量[34]。PXT3003是由低剂量的巴氯芬、山梨醇和纳曲酮组成的复方制剂,临床前研究结果表明该药可下调PMP22转基因大鼠PMP22蛋白的表达并改善髓鞘形成。PXT3003已完成法国多中心Ⅱ期和欧美多中心Ⅲ期临床研究,结果表明高剂量组相比安慰剂组能够改善CMT1A患者的部分疗效重点指标,且安全性较好[35, 36]。由于Ⅲ期临床研究中高剂量组制剂存在析晶问题,造成该组约半数患者破盲,美国食品药品监督管理局要求在欧洲和美国再增加一个Ⅲ期临床试验以验证PXT3003的有效性。目前,国内PXT3003治疗CMT1A患者的多中心、随机、双盲、安慰剂对照Ⅲ期临床试验正在开展。
CMT是最常见的周围神经遗传性疾病,具有显著的临床表型和遗传表型异质性。随着二代测序等基因检测技术的发展和普及,越来越多的CMT致病基因和新发突变位点被发现和克隆,极大地丰富了CMT的基因谱。康复训练治疗和外科矫形手术治疗仍是CMT的主要治疗手段,针对发病机制中重要靶点的多种特异性治疗药物的研究处于临床试验阶段或临床前研究阶段,有望将来为CMT的治疗带来新的突破。
姚晓黎, 何若洁. 腓骨肌萎缩症的诊治[J]. 中华神经科杂志, 2024, 57(3): 290-297. DOI: 10.3760/cma.j.cn113694-20230915-00165.
所有作者声明无利益冲突
None declared
1. 以下哪项是腓骨肌萎缩症(CMT)最常见的临床分型?
A. CMTX
B. CMT2B
C. CMT1A
D. CMT4C
2. 腓骨肌萎缩症与远端型肌营养不良均表现为四肢远端无力,肌萎缩逐渐向近端发展,以下哪项检查对于两者鉴别最重要?
A. 头颅磁共振成像
B. 肌电图
C. 脑电图
D. 脑脊液检查
3. 关于腓骨肌萎缩症下面哪项描述不正确?
A. 儿童或青少年期起病,缓慢进展
B. CMT1型发病年龄较早,CMT2型发病年龄较晚
C. 临床表现为双下肢远端肌肉萎缩,呈“倒香槟酒瓶”或“鹤腿”
D. 有锥体束征表现
4. 以下哪项是CMT最常见的致病基因?
A. PMP22
B. GJB1
C. GDAP1
D. MPZ
5. 以下哪项为CMT1的主要神经电生理特点?
A. 神经传导速度轻度减慢或正常,复合肌肉动作电位波幅降低
B. 神经传导速度显著减慢
C. 肌电图呈病理干扰相
D. 常见纤颤电位和正锐波

























