
血管免疫母细胞性T细胞淋巴瘤(AITL)是外周T细胞淋巴瘤的一种独特亚型,肿瘤细胞起源于滤泡辅助T细胞,背景细胞种类复杂,诊断相对困难,主要依靠组织病理学检查确诊。AITL具有恶性程度高、进展快、预后差等特点,目前仍缺乏有效的治疗手段。随着高通量测序的发展,现已发现表观遗传学(DNA甲基化修饰、组蛋白修饰)、T细胞受体及肿瘤微环境相关基因突变与AITL的发病机制及预后相关,为AITL潜在的靶向治疗提供了有力支撑。本文主要对AITL的分子特征及相关治疗的研究进展进行综述,以期帮助临床医生了解AITL基因组学研究进展,为个体化精准治疗提供依据。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
血管免疫母细胞性T细胞淋巴瘤(angioimmunoblastic T-cell lymphoma,AITL)起源于滤泡辅助T细胞(follicular helper T-cell,TFH),属于外周T细胞淋巴瘤(peripheral T-cell lymphoma,PTCL)亚型之一,具有独特的形态和复杂的生物学特性,目前主要的诊断方法是淋巴组织活检,临床上以化疗为主要的治疗方案,缓解率低,复发率高,5年总存活率为32%~44%[1]。
AITL的致病因素目前尚不明确,有研究表明其与EB病毒(EBV)感染密切相关,约97%的AITL患者早期即可发现EBV阳性的 B细胞,且在其邻近可发现TFH表型的肿瘤细胞,有人提出这两种细胞相互作用为肿瘤的形成提供了支持作用,但具体机制仍不明确[2]。AITL的肿瘤细胞较少,背景细胞复杂,肿瘤微环境(tumor microenvironment,TME)在促进肿瘤发生、进展和免疫逃逸过程中发挥着重要作用。此外,AITL的发生涉及多种基因突变,这些突变会对肿瘤T细胞及TME产生不同程度的影响,进而影响AITL的发生发展。AITL的发生机制及EBV感染和相关基因突变起的作用见图1。
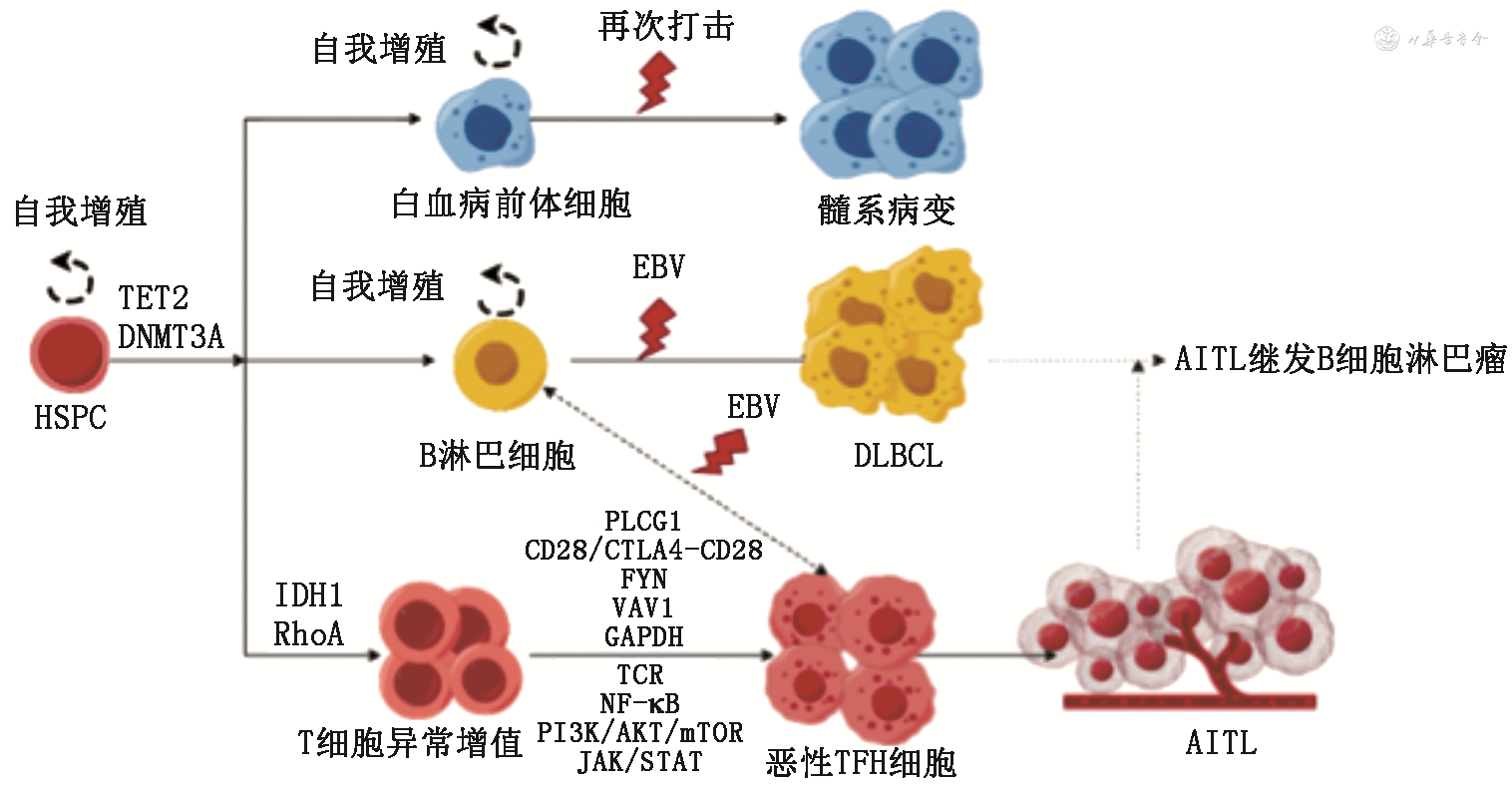
注:DNMT3A为DNA甲基转移酶3A;EBV为EB病毒;AITL为血管免疫母细胞性T细胞淋巴瘤;HSPC为造血干细胞/祖细胞;DLBCL为弥漫大B细胞淋巴瘤
1. DNA甲基化修饰:异常甲基化是血液系统恶性肿瘤发生的关键,对AITL患者样本的外显子组测序发现,DNA甲基化相关基因如:TET甲基胞嘧啶双加氧酶2(TET methylcytosine dioxygenase 2,TET2)、DNA甲基转移酶3A(DNA methyltransferase 3 alpha,DNMT3A)、异柠檬酸脱氢酶2(isocitrate dehydrogenase,IDH2)的突变普遍存在。(1)TET2突变:TET2编码双加氧酶,其突变后酶活性受损,导致哺乳动物基因组中5-甲基胞嘧啶氧化为5-羟甲基胞嘧啶的功能缺陷,诱导异常的DNA甲基化,其在AITL中的突变率约占80%,且超过一半的AITL中可发现多个TET2突变,但TET2突变发生在造血干细胞/祖细胞(hematopoietic stem/progenitor cell,HSPC)时并不直接导致AITL,当其与驱动疾病表型的继发性突变共同发生时才会导致恶性肿瘤的发生。研究表明,AITL患者中TET2突变与疾病晚期、血小板减少、高国际预后指数评分和较短的无进展生存期(PFS)及总生存期(OS)相关,且当TET2和TP53联合突变时,预后较单独TET2或TP53突变有所改善[3]。(2)DNMT3A突变:DNMT3A编码调节DNA甲基转移酶(DNMT)介导DNA甲基化,缺失后会阻碍HSPC分化,进而使骨髓中HSPC增多并恶性转化。28.21%的AITL中可发现DNMT3A突变,且与TET2和Ras同源家族成员A(ras homolog gene family member A,RhoA)发生共突变频率较高。AITL患者中DNMT3A表达相对较低,仅有少部分表达较高水平的DNMT3A,其中,低表达的患者更容易发生复发和转移,这可能与趋化因子信号传导、细胞因子受体相互作用、免疫IgA产生和细胞周期上调有关[4]。此外,DNMT3A突变可激活细胞毒性T细胞的转录,这可能与AITL患者较低的OS相关。(3)IDH2突变:IDH2编码异柠檬酸脱氢酶2,其突变影响参与DNA去甲基化和组蛋白去甲基化的相关酶以及TET2蛋白。约20%的AITL中可检测到IDH2突变,且其常与TET2共存。IDH2的突变使细胞拥有了TFH表型及独特的形态特征,并使T细胞受体(TCR)信号传导和T细胞分化的基因超甲基化增加[5]。Ye等[3]的研究显示IDH2突变会显著降低AITL患者的生存率,然而亦有研究提出IDH2R172突变与较好的PFS、OS以及诱导治疗反应相关[3,6]。
2.T细胞受体信号及下游信号通路:高达60%~70%的AITL中可发现RhoA突变,约50%的AITL中观察到其他与TCR信号相关的基因突变,如:PLCG1(14.1%)、CD28(9.4%)、CTNNB1(6%)、GTF2I(6%)、FYN(3%~4%)和VAV1(5%),这些突变与多种信号分子和激酶相互作用,激活TCR及下游信号通路,进而影响TFH的增殖和分化[7]。(1)RhoA突变:RhoA的突变率在AITL中仅次于TET2,且仅在AITL和非特异性外周T细胞淋巴瘤(PTCL-NOS)中被发现,具有特异性,其突变率与肿瘤细胞的比例相关。RhoA突变破坏了GTP的结合,从而导致RhoA信号通路的改变及生物学功能受损。最常检出的G17V突变通过加速TCR信号转导和增强可诱导共刺激分子(ICOS)-磷脂酰肌醇3-激酶(PI3K)信号通路,诱导TFH谱系分化并驱动AITL转化[8]。此外,RhoA突变还可诱导自身免疫,这或许与AITL的免疫失调相关。在AITL中还发现了其他活性(p.C16R和p.G14V)和非活性(p.G17E)RhoA突变,说明其可通过其他机制诱导T细胞淋巴瘤的发展[9]。然而,研究表明RhoA突变的AITL患者虽早期ECOG较低,但总体预后较好,本中心对20例AITL临床分析的结果亦支持上述观点,数据待发表。(2)其他TCR相关基因突变:PLCG1突变后可激活核因子κB(NF-κB)/活化T细胞核因子1(NFAT1)和干扰素调节因子4(IRF4),增强TCR信号从而促进AITL发展。CD28的特定突变(p.D124、p.T195)和融合(CTLA4-CD28、ICOS-CD28)是TFH淋巴瘤所特有的,表达CTLA4-CD28融合基因的小鼠出现淋巴结病变和脾肿大等炎症反应,寿命缩短,且其体内T细胞的激活水平升高,进一步分析显示CD4+T细胞中的基因表达模式与AITL非常相似,表明CD28突变在AITL的发生中起着重要作用[10]。FYN酪氨酸激酶亦在T细胞活化中起重要作用,FYN-TRAF3IP2融合基因在小鼠HSPC表达可导致TCR下游异常激活、NF-κB信号传导、T细胞恶性转化,已有研究在AITL样本中检测到该融合基因[11]。多功能蛋白VAV1与RhoA蛋白相似,可催化RHO GTPase中核苷酸的交换,还可与RhoA G17V特异性结合,通过磷酸化酪氨酸加速TCR信号传导[12]。既往研究通过构建小鼠模型发现VAV1获得性突变的表达有利于T细胞淋巴瘤的发生,CD4+T细胞中表达三价亚型VAV1突变体在Trp53缺失的情况下可以在小鼠体内触发AITL的发展,这可能与野生型VAV1在TFH细胞中的生理功能恶化有关[13, 14]。然而,上述突变中仅CD28突变可导致较低的PFS及OS,其他与TCR通路相关的基因突变与生存无关。
3.组蛋白修饰:组蛋白修饰参与染色质状态调节,有研究者在AITL血浆循环肿瘤DNA(ctDNA)中发现组蛋白修饰相关基因,如KMT2D(28.6%)和CREBBP发生了频繁的体细胞突变,并提出ctDNA的突变量与AITL的复发和进展显著相关,提示组蛋白修饰基因突变与AITL化学耐药及较短的PFS相关[15]。
4.其他:(1)CARD11突变:Caspase募集结构域蛋白11(CARD11)与免疫相关,参与NF-κB信号通路。CARD11突变并不足以导致淋巴瘤,但其可活化T细胞,导致调节性T细胞(Treg)、TFH和滤泡调节性T细胞(TFR)在体内过度积累,使ICOS、细胞毒性T淋巴细胞相关蛋白4(CTLA-4)和程序性死亡受体1(PD-1)分子表达水平升高,或与T细胞淋巴瘤的高侵略性相关。高达30%的PTCL中可发现CARD11获得性突变,但其在AITL中发生的频率较低,目前尚没有研究报道CARD11对T细胞的内在影响[16]。(2)BCL-6共抑制因子(BCOR)突变:BCOR可选择性地与BCL-6的POZ结构域相互作用,抑制转录。Tanaka 等[17]的研究首次证明BCOR 在 T 淋巴细胞恶性肿瘤的发病过程中起抑制作用。Kang等[18]在18例AITL样本中发现2例BCOR K607E突变(11.1%),突变型BCOR与BCL6、PCGF1和RING1B蛋白的亲和力较低,进而显著增强T细胞增殖、AKT磷酸化和白细胞介素(IL)-2的表达,上调HOX和S100蛋白基因的表达,但BCOR K607E突变与淋巴瘤患者的性别、年龄、病变部位及生存无关。(3)甘油醛3-磷酸脱氢酶(GAPDH)突变:Mondragón等[19]构建了过表达GAPDH的小鼠以评估体内代谢在T细胞成熟中的作用,这些小鼠最终出现了与AITL患者相同的临床特征和表型,表明T细胞中GAPDH的过表达可诱导TFH亚群中TCR的克隆并导致与肿瘤细胞生长相关的突变;此外,其过表达还可以使非典型NF-κB通路上调[19]。
除上述提到的基因突变外,有学者通过检验外周血中游离DNA(cfDNA)将KDM5A、STAT1、FANCM、ERBB4、PIK3R5和 NSD1定义为AITL新的复发性突变,为AITL的研究提供新的方向[20]。
Scourzic等[21]构建的DNMT3AR882H Tet2-/-小鼠模型证明了小鼠HSPC中TET2缺失和DNMT3AR882H表达会导致髓系和淋巴系统恶性肿瘤,最终约有60%小鼠出现AITL样疾病,证明了TET2和DNMT3A是驱动AITL形成的第一步。Zheng 等[4]的研究进一步表明DNMT3AR882H 突变可以加速TET2-/-小鼠AITL的发展,这可能是DNMT3A突变增强了PD-1/程序性死亡受体-配体1(PD-L1)、ICOS/可诱导共刺激分子配体(ICOSL)、CD28/CD86和细胞间黏附分子-1(ICAM1)/整合素αL((ITGAL)的相互作用,从而刺激TFH扩增。Zang等[22]通过构建TET2-/-RhoAG17V的小鼠发现TET2缺失和RhoA突变可通过FoxO1失活破坏正常CD4+T细胞功能,进而导致细胞增殖增加、凋亡或死亡减少,TCR信号异常激活,以及辅助性T细胞17(Th17)、Treg细胞和TFH细胞之间的失衡,从而诱导类似于AITL患者的炎症表现[22]。Leca 等建立了TET2和IDH2共突变的小鼠模型,发现双突变小鼠脾脏中CD4+CXCR5+PD-1+T细胞表达ICOS的比例增加,提示TFH的扩增,而携带双突变的TFH与生发中心B细胞的串扰发生改变,促进B细胞克隆扩增,同时减少Fas-FasL相互作用,减少B细胞凋亡,阐述了AITL患者并发B细胞异常增殖的机制[5]。
AITL的TME具有异质性,其特点是肿瘤T细胞与浸润性B细胞(多数EBV阳性)共存,除此之外还包括非肿瘤性T细胞、滤泡树突状细胞(FDC)、巨噬细胞以及高内皮小静脉(HEV)、嗜酸性粒细胞及各种细胞因子。
1. EBV感染B细胞:EBV 感染与 AITL 中 B 细胞的克隆扩增相关,最终可能导致并发弥漫大 B 细胞淋巴瘤(DLBCL)。据报道,大多数AITL 病例中其EBV+B 细胞因破坏性高突变而失去功能性 Ig,并不需要 B 细胞受体(BCR)信号。在新发或与 AITL 相关的DLBCL中,TET2和DNMT3A突变是唯一与EBV阳性呈显著正相关的基因突变,推测 TET2和 DNMT3A突变与 EBV 共同作用于 DLBCL 的发展。伴TET2突变的B细胞与TFH相互作用,可促进生发中心B细胞扩增,从而使滤泡结构发生破坏。另外,在AITL的CD19+B细胞发现 NOTCH1 突变,或可增强 TET2 突变效应。携带TET2和NOTCH1突变的B细胞与携带TET2和RHOA突变的TFH细胞之间的相互作用可能介导AITL发育所必需的双向信号传导[23]。而具体针对AITL中EBV基因组学分析,Bahri 等[24]发现EBV多数处于潜伏状态且具有克隆性,根据突变基因可大致分为两株,其中BRLF1 S542N/BZLF1 A206S 突变病毒株与患者生存期短相关。在AITL中变异最多的潜伏基因是 EBNA-1、EBNA-2 和 EBNA-LP,这些因素都支持 EBV 在 AITL 中的作用[24]。另外,他们还通过研究AITL 中的 EBV 转录组学,发现AITL 的潜伏期为 Ⅱc(有 BNLF2a 基因表达),Bam-HI A 向右转录本(BARTs)是在 AITLs中表达最多的潜伏转录本,编码人类白细胞介素 10(vIL-10)同源蛋白的 BCRF1 也有表达,这种共同表达可能有助于免疫逃逸和感染细胞的存活。基于此,可以推测EBV 在 AITL中起着致病作用[25]。
2. 非肿瘤T细胞:TET2 突变在启动 AITL 中除了能增加TFH 细胞数量外,还能破坏FOXP3的稳定性导致 Tregs减少,并增加Th17的数量,参与免疫高反应性。另外,通过多组学分析还发现AITL TME 中,B 细胞中CD73 和 CXCR5标记物减少,扩增的CD8+T 细胞表型衰竭(表现为功能障碍、细胞毒性受损以及趋化因子 XCL2 和 XCL1 上调)。相对新诊断AITL,复发难治AITL(RR-AITL)患者中TME具有显著差异,表现为CD8+T细胞比例降低,Treg比例升高,T细胞耗竭,而 B 细胞的干性增强,并表现出明显的恶性特征[26]。
3. FDC、HEV及细胞因子:FDC和HEV的增加是AITL所特有的,FDC位于AITL克隆附近,与TFH相互作用可产生丰富的IL-21,IL-21可通过激活STAT3通路诱导B细胞增殖、抗体分泌、浆细胞分化和凋亡;此外,IL-21在T细胞中的表达增加,会使B细胞失调及Th17细胞进一步积累,导致自身免疫性疾病,这可能与AITL患者的多克隆高γ球蛋白血症和免疫缺陷有关[27]。而血管内皮生长因子的过度分泌可能与 HEV 的增加有关,但 HEV 如何影响 AITL 的建立尚不清楚[23]。有研究提出IDH2突变可通过增加浆细胞数目和HEV来影响AITL的TME[5]。此外,肿瘤性巨噬细胞通过产生细胞因子、生长因子和抑制免疫检查点来产生免疫抑制性TME,导致肿瘤的发生发展、免疫调节和远处转移,其与患者的OS密切相关。TET2突变对细胞因子的影响目前仍存在争议,有研究表明其可增加IL-17的分泌进而引起炎症性微环境,亦有研究表示其会抑制Th1和Th17极化以及相关细胞因子的产生[28]。
目前,AITL的一线治疗仍是以蒽环类药物为基础的化疗,大多数患者在经过一线治疗后仍表现出缓解率低、进展快、复发率高。与基因突变相关的靶向治疗为AITL的治疗提供了新策略,有望改善AITL患者的预后。
近年来,新药治疗AITL取得了良好的进展,表观遗传学失调在AITL的发生中起着重要作用,针对表观遗传学失调的药物,如去甲基化药物、组蛋白去乙酰化酶抑制剂在临床上得到了广泛的应用。研究表明,这些药物可以作为补救性方案治疗R/R AITL以及高龄患者,且可以明显改善预后[29, 30, 31]。信号通路抑制剂可以通过阻断TCR信号传导,抑制NF-κB通路、JAK1/2通路及PI3K通路等使AITL患者受益[32, 33, 34]。除上述提到的基因突变相关药物,单克隆抗体、免疫检测点抑制剂、抗叶酸药物、嘌呤核苷磷酸化酶抑制剂、新型靶向线粒体等药物也在临床上得以应用。此外,许多与TET2功能相关的靶向药物已进入临床前研究,有望进一步丰富临床用药。
AITL的基因组学研究及治疗进展,阐述其表观遗传失调、组蛋白修饰、TCR及其下游信号通路相关的突变在AITL发病机制中的作用及基因突变相关药物在AITL患者中的应用,以期通过靶向治疗改善AITL患者的预后。
左素琼, 周盼, 朱尊民. 血管免疫母细胞性T细胞淋巴瘤基因组学研究及治疗进展[J]. 中华医学杂志, 2024, 104(10): 783-787. DOI: 10.3760/cma.j.cn112137-20231104-01008.
所有作者声明不存在利益冲突

























