
本文报道了1例先天性鼻及鼻窦发育畸形的患儿。患儿女,14岁,因“自幼外鼻畸形,双侧鼻塞”入住北京同仁医院耳鼻咽喉头颈外科。患儿出生后出现喘憋,吃奶时呛咳,于我科诊断为鼻畸形、后鼻孔闭锁。术前对患儿全血进行全外显子基因检测。于全身麻醉下行鼻内镜下右鼻膜性后鼻孔闭锁开放术,术后效果良好。本文讨论了先天性鼻畸形的分类、发育畸形的原因及先天性后鼻孔闭锁的处理。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
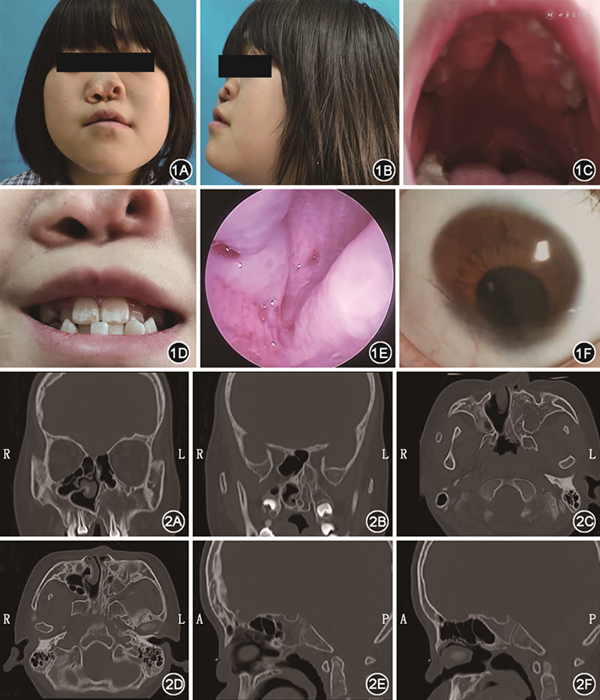
患儿女,14岁,因“自幼外鼻畸形、双侧鼻塞”入院。患儿出生后出现喘憋,吃奶时呛咳,自幼经口呼吸,无嗅觉,发音时有闭合性鼻音,合并右眼外观畸形,听力语言发育正常,无智力障碍,于2021年11月18日以“先天性鼻畸形、先天性眼畸形”收入我科。既往先天性室间隔缺损,10年前行手术修补,无头面部畸形家族史。体格检查:外鼻畸形,鼻背塌陷,鼻小柱缺失,左侧前鼻孔闭锁。口腔检查:硬腭高拱,硬腭裂,牙列不齐(图1A~D)。鼻内镜检查:左侧鼻腔内结构无法窥及,右侧下鼻甲及中鼻甲结构异常,右侧后鼻孔闭锁(图1E)。眼距宽,右眼上睑下垂,右侧瞳孔位置偏下(图1F),右眼直接、间接对光反射不灵敏,右眼视力差,左眼视力正常;听力正常。术前鼻窦CT:左侧上颌窦、鼻腔及鼻甲未发育,右侧中鼻甲未发育(图2A、D),左侧后鼻孔骨性闭锁(图2B、C),双侧鼻骨塌陷,鼻咽顶后壁软组织增厚,右侧后鼻孔膜性闭锁(图2C、F),左侧后鼻孔骨性闭锁(图2D、E)。初步诊断:鼻畸形,鼻窦畸形,后鼻孔闭锁,眼畸形,心脏术后。
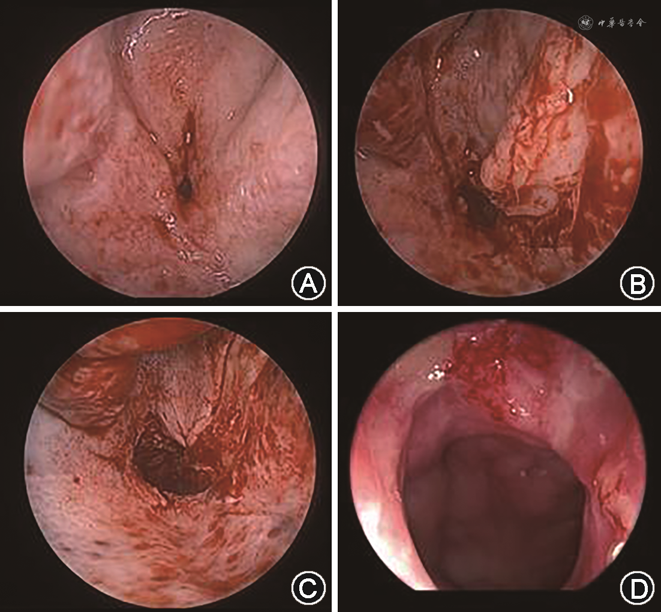
鉴于其家属仅要求解决鼻腔通气而非鼻外观问题,患儿于全身麻醉下接受鼻内镜下右侧膜性后鼻孔闭锁开放术。术中见右侧后鼻孔膜性闭锁,使用动力系统切除膜性后鼻孔黏膜至后鼻孔骨缘(图3A),使用剥离子沿骨面向前向上剥离黏膜,掀起并保留黏膜瓣。使用骨钻磨除骨性鼻中隔后部及骨性后鼻孔上缘(图3B),磨至后鼻孔宽度满意(图3C),复原黏膜瓣,鼻腔放置硅胶鼻导管局部压迫。术后2 d取出鼻导管,鼻腔无明显活动性出血,黏膜对位良好。术后2个月复查,见鼻内镜下右侧后鼻孔通畅(图3D),患儿及家属对疗效满意。
由于先天性颅面部畸形常伴有基因突变,术前经患儿家长知情同意后,抽取患儿全血进行全外显子基因检测。未发现临床表型高度相关且致病性证据充分的点突变或大片段缺失重复,其他可疑基因变异见表1:发现潜在转化生长因子β结合蛋白3(latent TGF-beta binding protein 3,LTBP3)和赖氨酸乙酰转移酶6(lysine acetyltransferase 6,KAT6A)的突变。
先天性鼻及鼻窦发育畸形患儿可疑基因变异
先天性鼻及鼻窦发育畸形患儿可疑基因变异
| 基因 | 基因全称 | 染色体位置 | 转录本外显子 | 核苷酸氨基酸 | 纯合/杂合突变 | 疾病/表型(遗传方式) |
|---|---|---|---|---|---|---|
| LTBP 3 | latent TGF-beta binding protein-3 | chr11:653 10639 | NM_001130144;exo n18 | c.2533A>G(p.N845D) | 杂合 | 1.选择性牙齿发育不全6型(AR) 2.Geleophysic发育不良3型(AD) |
| KAT6 A | lysine acetyltransferase 6 | chr8:4178 9926 | NM_006766;exon17 | c.5812A>G(p.S1938G) | 杂合 | Arboleda-Tham综合症(AD) |
注:AR为隐性遗传;AD为显性遗传
先天性鼻畸形是一组在胚胎发育过程中形成的鼻部结构异常,其发病率在不同地区和人群之间有所变化。研究表明,先天性鼻畸形的发病率通常较低,约为出生人口的1/40 000~1/20 000[1]。其发病可能与复杂的胚胎发育过程以及多基因和环境因素的相互作用有关。根据临床特征,先天性鼻畸形可分为以下4类:发育不全(hypoplasia and atrophy)、增生畸形(hyperplasia and duplications)、裂(clefts)和肿瘤及血管异常(neoplasms and vascular anomalies)[2]。这些分类有助于更好地理解畸形的类型和特点,从而为诊断和治疗提供指导。该患者属于第一类发育不全。
在胚胎发育过程中,鼻的形成涉及复杂的组织胚胎学过程,胚胎中的不同细胞类型需要准确分化和定位形成正常的鼻部结构[3]。先天性鼻畸形可能是多种因素作用的结果,包括遗传突变、环境暴露、母体健康状况等,一些畸形可能由于胚胎发育过程中细胞迁移、增殖或分化时发生异常所致[4]。CT影像对鼻部异常及其相关的颅骨病变具有重要提示作用,有助于手术方案的设计[5]。本例患者行全血基因检测时发现LTBP3和KAT6A基因突变。LTBP3突变与牙齿异常和身材矮小综合征相关,其特征为下颌缺损、上颌骨发育不全、身材矮小、短肌缺损、动脉瘤和胸主动脉夹层[6]。KAT6A综合征患者的面部特征是鼻尖较宽、上唇较薄,随着年龄增长可能更为明显,其他常见的面部特征包括双颞狭窄、鼻梁突出和鼻中部短平[7]。KAT6A基因突变在表型上存在一定差异,与发育迟缓有关,曾有病历报道其可能导致房间隔缺损[8]。这些可疑变异的临床意义尚不明确,需进一步研究以明确其与受检者临床表型的相关性。
先天性后鼻孔闭锁最早于1755年由罗德尔描述,是新生儿后鼻孔狭窄或闭塞的一种病理状态[9]。据报道,其发生率为1/7 000~1/5 000,女婴患病率高于男婴[10, 11]。后鼻孔闭锁根据组织类型分为膜性、骨性和混合型闭锁,其中单侧闭锁占60%,双侧占40%[12]。后鼻孔闭锁可能导致新生儿无法通过鼻腔呼吸,情况危急时需紧急处理。如患儿幸存并进入幼儿期,可能需要进一步手术治疗,以确保正常的鼻部功能发展。本例患儿为混合型后鼻孔闭锁,家长述患儿出生后只能经口呼吸,但因尚未明显影响生长发育,14岁后为尝试通过手术实现经鼻呼吸而就诊。由于患儿未成年,家长仅要求改善通气功能。由于其左侧鼻腔未发育,完全骨化闭锁,无手术可能性,遂开放右侧膜性闭锁以改善通气功能,未行鼻窦畸形矫正手术。
综上所述,先天性鼻畸形可能导致鼻腔大小、形状或位置异常,可能影响呼吸、声音传导以及与鼻腔相关的其他功能。对于鼻畸形的理解有助于提高早期诊断和治疗效果,从而改善患者的生活质量。未来的研究可以进一步探索不同基因变异、环境因素以及胚胎发育过程中的相互作用,以更全面地解析先天性鼻畸形的形成机制。
玄丽佳, 庄梦岩, 赵亚娟, 等. 先天性鼻及鼻窦发育畸形1例[J]. 中华耳鼻咽喉头颈外科杂志, 2024, 59(3): 253-255. DOI: 10.3760/cma.j.cn115330-20231022-00163.
所有作者声明无利益冲突

























