
尼曼-匹克病(Niemann-Pick disease,NPD)属于常染色体隐性遗传病,是一种罕见的糖脂代谢异常疾病[1]。根据临床症状及基因突变的不同,可分为3种类型,其中A型和B型是由于SMPD1 基因突变导致其编码的酸性鞘磷脂酶(acid sphingomyelinase,ASM)缺乏,引起神经鞘磷脂代谢障碍,使得后者在单核-巨噬细胞内蓄积,从而出现肝脾肿大等症状[2]。C型是由于NPC1或NPC2基因突变导致胆固醇转运障碍,从而在细胞内大量沉积[3]。临床上以A型多见,A型与B型的主要区别在于是否有神经系统受累,B型患者一般智力正常,发病晚,症 状轻,病程进展慢[4]。为了提高对本病的认识及诊断准确率,现对我院5年来确诊的2例NPD的临床资料进行回顾性分析,并复习相关文献。
版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
尼曼-匹克病(Niemann-Pick disease,NPD)属于常染色体隐性遗传病,是一种罕见的糖脂代谢异常疾病[1]。根据临床症状及基因突变的不同,可分为3种类型,其中A型和B型是由于SMPD1 基因突变导致其编码的酸性鞘磷脂酶(acid sphingomyelinase,ASM)缺乏,引起神经鞘磷脂代谢障碍,使得后者在单核-巨噬细胞内蓄积,从而出现肝脾肿大等症状[2]。C型是由于NPC1或NPC2基因突变导致胆固醇转运障碍,从而在细胞内大量沉积[3]。临床上以A型多见,A型与B型的主要区别在于是否有神经系统受累,B型患者一般智力正常,发病晚,症 状轻,病程进展慢[4]。为了提高对本病的认识及诊断准确率,现对我院5年来确诊的2例NPD的临床资料进行回顾性分析,并复习相关文献。
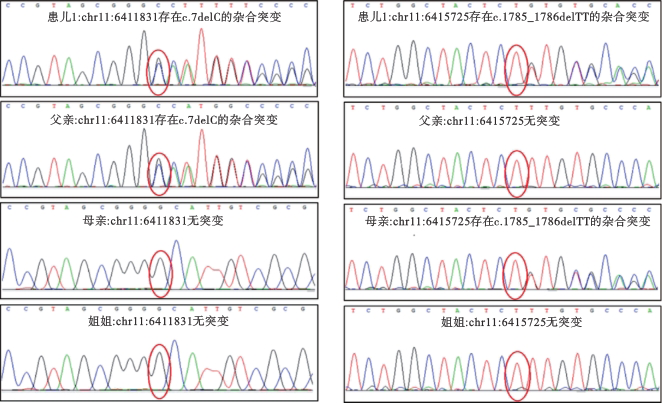
患儿,女,10月龄,20余天前因发热就诊于当地医院,给予抗细菌、抗病毒治疗,血常规提示小细胞低色素性贫血,予补充铁剂“右旋糖酐铁”治疗。口服铁剂16 d后患儿血红蛋白进一步下降,为进一步治疗就诊于我院,门诊以“贫血待查”收住院。体格检查:发育正常,神志清楚,精神一般,有轻度贫血貌,唇欠红润,咽稍充血。右侧腹股沟区触及到直径4 mm左右的淋巴结,质软,活动度尚可。腹软,肝肋下6 cm,质稍韧,脾肋下5 cm,质软。四肢无畸形,关节无红肿,运动无障碍,肌张力、肌力正常。血常规:白细胞总数5.30×109/L,中性粒细胞计数4.40×109/L,血小板计数114×109/L,血红蛋白68 g/L,MCV:69.0 fl,C反应蛋白<5.00 mg/L;肝功能:谷丙转氨酶99.50 U/L,谷草转氨酶203.21 U/L,总胆红素34.17 μmol/L,直接胆红素26.49 μmol/L;血脂:甘油三酯1.96 mmol/L,高密度脂蛋白0.45 mmol/L。胸部CT:肺部纹理增多,局部稍模糊,左肺可见少许斑点状致密影,考虑为支气管炎。B超:肝右叶肋下33 mm,肝脏边缘规则,肝内回声均匀,未见实性光团;脾脏大小130 mm×35 mm,肋下55 mm,边缘规则,其内回声均匀,提示肝、脾增大。骨髓细胞形态学检查:红系增生,尼曼-匹克样细胞多见(图1)。进一步完善基因检测结果显示患儿在NPD相关基因SMPD1存在两处杂合突变c.7delC(缺失突变)和c.1785_1786delTT(缺失突变),经家系验证显示分别来自父亲和母亲,患儿姐姐两个位点均无突变(图2)。这两处突变在HGMD 数据库中均有致病报道,报道疾病为NPD。综上结果该患儿诊断为NPD A型。给予保肝及其他对症治疗。一年后患者因重症肺炎再次入院,此时患者出现脾功能亢进、肝功能损害、贫血、低白蛋白血症,对症治疗后好转出院。电话随访患者于2岁3月龄时因肺部感染死亡。
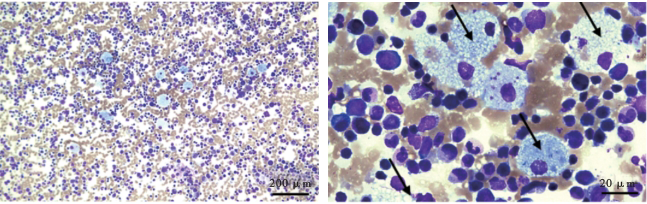
箭头所指为尼曼-匹克细胞
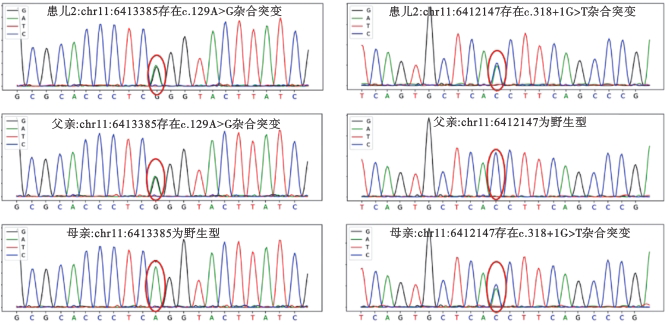
患儿,女,3岁,因入园前体检诊断为“发育迟缓”就诊于我院。患儿足月出生,6月龄能抬头,8月龄能独坐,1岁会叫爸爸妈妈,2岁会独走,现不会自己进食,不会跑、下蹲、双脚跳及单脚跳,社交能力差。父母身体健康,7岁哥哥生长发育正常。既往史:出生至今间断腹泻,为稀水样便,蒙脱石散、益生菌治疗后可好转,但停药后腹泻易反复;1.5岁时曾诊断贫血。体格检查:发育落后,神情略呆滞,精神尚可,颜面部粗糙、多毛,颜面及双手指端稍肿胀,双手掌侧皮肤可见类肝掌样改变,咽充血,双侧扁桃体Ⅱ°肿大,双肺听诊呼吸音粗。腹软,肝肋下6 cm,质韧,脾肋下11 cm,质硬。肌力、肌张力正常。眼底照相双眼底黄斑区可见樱桃红斑。血常规:白细胞总数4.28×109/L,中性粒细胞计数0.78×109/L,血小板计数94×109/L,血红蛋白115 g/L,MCV:80.8 fl。肝功能:谷丙转氨酶75.70 U/L;血脂:总胆固醇4.40 mmol/L,甘油三酯2.84 mmol/L,高密度脂蛋白0.41 mmol/L。B超:肝肋下30 mm,边缘规则,肝内回声增粗;脾大小120 mm×50 mm,脾肋下161 mm,边缘规则,提示肝、脾增大。胸部CT:双肺弥漫间质增厚网结影,双下肺为著。骨髓细胞形态学检查:骨髓增生活跃,粒系增生降低,红系增生活跃,血小板少见,尼曼-匹克细胞多见,细胞体积较大且胞质中含大量空泡,少量细胞吞噬成熟红细胞及血小板(图3)。进一步完善基因检测结果显示患儿在SMPD1基因,染色体位置chr11:6413385存在一处杂合突变c.129A>G,该突变来自于父亲,母亲为野生型。在SMPD1,染色体位置chr11:6412147存在一处杂合突变c.318+1G>T,该突变来自于母亲,父亲为野生型,家系验证为复合杂合突变(图4)。验证位点1 HGMD 数据库未收录,验证位点2 HGMD 数据库有致病报道,报道疾病为NPD。患儿骨髓细胞中可见典型的尼曼-匹克细胞,且肝脾肿大,伴有血小板低下,神经系统受累,眼底可见樱桃红斑,肺部有纤维化,结合患儿智力发育低下,提示为NPD A型。考虑患儿一般情况尚可,准予出院观察,嘱家长出院后加强护理,定期监测血常规、血脂、胸部CT及肺功能等。一年半后患者因全身浮肿再次入院,此时患者出现大量腹水、弥散性血管内凝血、呼吸道感染、肝功能衰竭、脾功能亢进、低蛋白血症、低钾血症、血小板减少、贫血症状,家长放弃治疗出院。
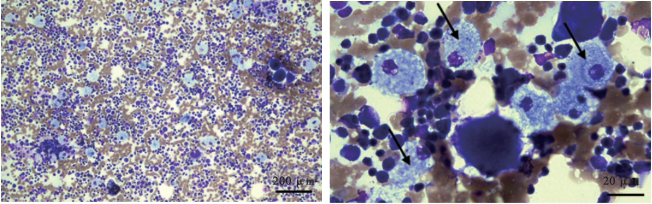
箭头所指为尼曼-匹克细胞
A型NPD又称急性神经型或婴儿型,该类型发病率最高,约为85%,严重程度也最高,多在出生后2~4个月内出现肝脾肿大,少数在出生后几周或1岁后发病,表现为消瘦、快速进展的神经系统退行性病变、智力及运动能力减退、肌张力低、黄疸,约半数有眼底樱桃红斑,所有婴儿都出现进行性限制性呼吸道症状,并伴有继发吸入性呼吸道感染,多因呼吸衰竭死亡[5]。本文2例患儿均出现肝脾肿大,骨髓中均见泡沫状尼曼-匹克细胞,血象有不同程度改变,血小板减少,均出现贫血症状。病例1早期发育正常,病例2发育迟缓,2例患儿的肌张力均正常。2例患儿均检测到SMPD1的突变,病例1两处突变均为碱基缺失造成的移码突变,对蛋白质的影响较大。病例2的两处突变均为错义突变,其中一处突变未在HGMD数据库中收录。2例患儿预后均较差,临床表现和实验室检查结果均是典型的A型NPD特征。国内一项大样本的研究发现,外周血白细胞残留ASM活性和血浆7‐KC水平与NPD临床表型之间存在显著相关性,表明这些生化参数对SMPD1新变异的预测和解释具有重要意义[2]。由于本院未开展ASM检测项目,所以2例患儿的ASM水平未知。
目前NPD尚无有效治疗方案,一般以对症支持治疗为主。严重脾功能障碍的患儿,可通过部分脾切除术改善症状。对于严重肝功能及肺功能障碍的患儿可行肝移植。Liu等[6]报道7例接受肝移植的B型NPD患儿,移植后3周肝功能、血脂水平恢复正常,肺部症状也得到了明显改善,所有患儿均出现追赶生长。国内个案报道对患儿进行异基因造血干细胞移植后症状也可得到改善,但远期疗效仍待研究[7, 8]。对于慢性非神经性NPD(NPD B型)研究发现有望行神经鞘磷脂酶替代治疗[9, 10, 11],但尚未批准上市;美格鲁特可以改善C型NPD(NPC)患者的脂质运输缺陷,减少细胞内脂质的储积,成为治疗NPC进行性神经症状的首个药物[12, 13],但目前尚未有A型NPD临床治疗研究的报道。
总之,NPD是一种罕见的遗传性代谢疾病,临床上容易出现漏诊误诊的情况。如发现肝脾肿大,肝功能及血脂异常的患者,应尽早行骨髓形态学及基因学检测,尽早确诊。该病治疗难、预后差,对于有先证者家族及时进行产前基因检测及遗传咨询,对预防并降低 NPD的发病率具有重要意义。
所有作者均声明不存在利益冲突

























