
听神经病是以听力下降、言语识别困难为表现的一类特殊的听觉障碍疾病,病因机制复杂、临床诊疗仍有挑战。近年来,包括基因治疗在内的生物学治疗技术在耳聋治疗研究中展示出良好效果。本文总结听神经病基因治疗研究现状,以期推动听神经病的精准治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,每年从第1期至第12期共刊发10篇以上继教文章,文后附5道选择题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。
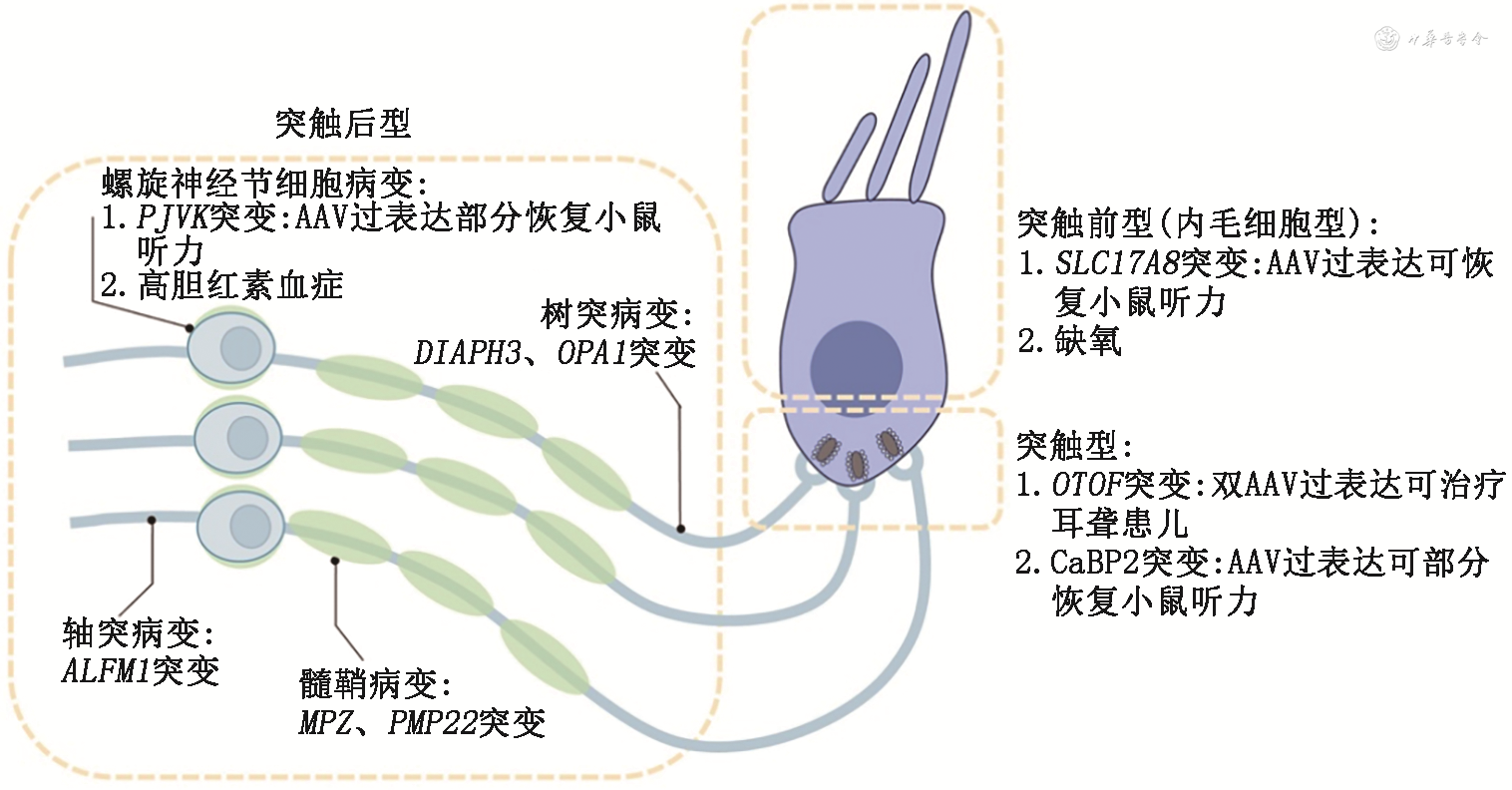
听觉信号传导复杂且高度精密,由内耳毛细胞、螺旋神经节、听神经共同完成声音从机械信号到神经编码的转导传递,最终在听觉中枢感知为听觉。从毛细胞到听神经环路中的病变均能导致感音神经性听力损失(sensorineural hearing loss,SNHL)。诊疗技术的发展推动SNHL分型的精细化,1992年顾瑞和于黎明[1]报道中枢性低频感音神经性听力损失病例。Starr等[2]于1996年发现10例具有相似听力学测试结果的病例:听性脑干反应(auditory brainstem response,ABR)严重异常或缺失,而耳声发射(otoacoustic emissions,OAE)和/或耳蜗微音器电位(cochlear microphonics,CM)可被引出,提示外毛细胞功能正常。Starr团队认为病因可能在于听觉通路同步不良,将病变定位在听神经并命名为听神经病(auditory neuropathy,AN)[2]。王秋菊教授总结听神经病这一类特殊的SNHL的临床表现:患者听觉信号时阈处理功能不足、有与纯音测听下降程度不符的言语识别困难,也称为听神经病谱系障碍(auditory neuropathy spectrum disorder,ANSD)[3]。我国2022年制定听神经病临床实践指南(2022版),定义听神经病为内毛细胞、突触、螺旋神经节细胞和/或听神经自身功能不良所致的听觉信息处理障碍性疾病[4]。得益于分子生物学以及遗传学技术发展,目前对于AN的病因及病变部位分型已更为深入。就病因划分,主要可分为环境因素和遗传因素2类[5]。导致AN的环境因素主要包括高胆红素血症、缺氧、维生素B1缺乏、噪声暴露等[6, 7, 8, 9]。遗传性AN分子机制复杂,目前已知SLC17A8、OTOF、CaBP2、PJVK、DIAPH3、AIFM1、OPA1、MPZ及PMP22等基因突变可导致AN[10]。根据环境因素、致病基因累及听觉环路中的部位,AN可分为突触前型(累及内毛细胞)、突触型(累及内毛细胞带状突触)和突触后型[4]。
因AN病因的复杂多样性以及病情严重程度不同,AN患者需要制定个体化的综合治疗方案。目前AN患者的临床治疗手段主要包括佩戴助听器和人工耳蜗植入[11]。助听器能够对声音进行有效放大,适用于轻中度聋患者。佩戴助听器后,AN患儿听力和言语功能改善[12],与匹配组SNHL患儿的语言发育情况相当[13, 14],但部分患儿语言发育仍比正常听力儿童迟缓[14];在噪声环境中,佩戴助听器的AN患儿的言语识别能力较匹配组SNHL患者更差[15]。总之,佩戴助听器不改变AN患者声音处理不同步的问题,在复杂声音环境中,助听器对言语识别功能的辅助效果尚未明确。对重度至极重度AN患者,人工耳蜗植入是目前临床治疗的首选。人工耳蜗植入设备可将声音信号转化为编码电信号,从而绕过受损的内耳毛细胞,直接刺激耳蜗螺旋神经元[16],该信号刺激被认为能够改善AN的听觉信号不同步问题[17]。人工耳蜗植入的治疗效果有赖于听神经信号传导功能的完整性,与病变位置及耳聋突变基因有着密切相关性[18]。目前认为突触前型和突触型的AN患者在植入后可获得与其他SNHL患者相当的听力及语言恢复效果,而对于突触后型AN患者,部分患者在人工耳蜗植入术后听力恢复效果不佳[18, 19, 20]。AN病因复杂,助听器及人工耳蜗植入治疗效果有限。
基因治疗利用过表达正常基因或抑制、修改有缺陷的基因来实现治愈疾病或提高抵抗疾病能力的目的[21],目前主要的基因治疗手段包括基因过表达、基因编辑、RNA干扰等。随着基因治疗工具效率和靶向性的提高,包括基因治疗在内的生物治疗技术已经在SNHL模式小鼠治疗中取得很好的治疗效果[22, 23, 24, 25, 26, 27, 28, 29]。不止于动物研究,基于腺相关病毒(adeno-associated virus,AAV)的遗传性耳聋基因治疗已开始进入临床试验,基因治疗有望作为未来遗传性AN临床治疗的一种新策略。目前非综合征型AN基因治疗以过表达策略为主;综合征型AN由于缺乏模拟人类耳聋表型的动物模型而未有突破,但其他系统基因治疗的成功有望为AN治疗提供新方案。本文梳理与AN相关的基因治疗的研究报道,以期探寻AN治疗新手段(图1)。
基因过表达是基因治疗的传统治疗策略,通过将目的基因构建到载体(通常是病毒),导入靶细胞实现外源性基因的表达,通过替代治疗来实现功能的恢复,适用于隐性纯合突变或显性负效应导致的遗传性疾病。由AAV介导的基因替代疗法已被应用于遗传性视网膜疾病、β-地中海贫血、杜氏肌营养不良的治疗,多项治疗药物获得美国食品和药物监督管理局(FDA)的批准上市[30, 31, 32]。利用基因过表达是耳聋基因治疗最有效的手段,在非综合征型AN的治疗研究中展示出良好效果(表1)。
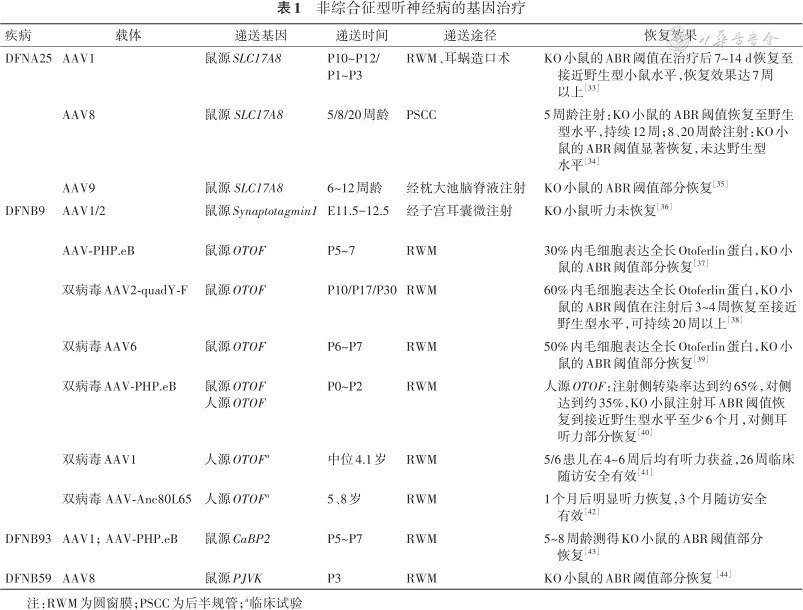
非综合征型听神经病的基因治疗
非综合征型听神经病的基因治疗
| 疾病 | 载体 | 递送基因 | 递送时间 | 递送途径 | 恢复效果 |
|---|---|---|---|---|---|
| DFNA25 | AAV1 | 鼠源SLC17A8 | P10~P12/ P1~P3 | RWM、耳蜗造口术 | KO小鼠的ABR阈值在治疗后7~14 d恢复至接近野生型小鼠水平,恢复效果达7周以上[33] |
| AAV8 | 鼠源 SLC17A8 | 5/8/20周龄 | PSCC | 5周龄注射:KO小鼠的ABR阈值恢复至野生型水平,持续12周;8、20周龄注射:KO小鼠的ABR阈值显著恢复,未达野生型水平[34] | |
| AAV9 | 鼠源 SLC17A8 | 6~12周龄 | 经枕大池脑脊液注射 | KO小鼠的ABR阈值部分恢复[35] | |
| DFNB9 | AAV1/2 | 鼠源Synaptotagmin1 | E11.5-12.5 | 经子宫耳囊微注射 | KO小鼠听力未恢复[36] |
| AAV-PHP.eB | 鼠源OTOF | P5~7 | RWM | 30%内毛细胞表达全长Otoferlin蛋白,KO小鼠的ABR阈值部分恢复[37] | |
| 双病毒AAV2-quadY-F | 鼠源OTOF | P10/P17/P30 | RWM | 60%内毛细胞表达全长Otoferlin蛋白,KO小鼠的ABR阈值在注射后3~4周恢复至接近野生型水平,可持续20周以上[38] | |
| 双病毒AAV6 | 鼠源OTOF | P6~P7 | RWM | 50%内毛细胞表达全长Otoferlin蛋白,KO小鼠的ABR阈值部分恢复[39] | |
| 双病毒AAV-PHP.eB | 鼠源OTOF 人源OTOF | P0~P2 | RWM | 人源OTOF:注射侧转染率达到约65%,对侧达到约35%,KO小鼠注射耳ABR阈值恢复到接近野生型水平至少6个月,对侧耳听力部分恢复[40] | |
| 双病毒AAV1 | 人源OTOFa | 中位4.1岁 | RWM | 5/6患儿在4~6周后均有听力获益,26周临床随访安全有效[41] | |
| 双病毒 AAV-Anc80L65 | 人源OTOFa | 5、8岁 | RWM | 1个月后明显听力恢复,3个月随访安全有效[42] | |
| DFNB93 | AAV1;AAV-PHP.eB | 鼠源CaBP2 | P5~P7 | RWM | 5~8周龄测得KO小鼠的ABR阈值部分恢复[43] |
| DFNB59 | AAV8 | 鼠源PJVK | P3 | RWM | KO小鼠的ABR阈值部分恢复 [44] |
注:RWM为圆窗膜;PSCC为后半规管;a临床试验
1. SLC17A8基因:SLC17A8基因编码VGLUT3蛋白,介导内毛细胞带状突触摄取谷氨酸囊泡,对听觉通路的发育及编码功能有着重要作用,突变导致突触间隙谷氨酸水平过低,动作电位难以产生,无法传递听觉信号,引起常染色体显性遗传非综合征型耳聋DFNA25,临床表现为渐进性高频耳聋[45, 46]。
在Vglut3基因敲除小鼠中可观察到正常的带状突触形态结构及突触囊泡循环,但给予小鼠声刺激后无法检测到ABR及蜗神经动作电位,2周龄后可见螺旋神经元及传入、传出末梢数量减少[45],此外小鼠表现出原发性癫痫伴有持续的运动行为异常[46]。Akil等[33]在新生Vglut3基因敲除小鼠模型中使用AAV1递送Vglut3基因可在内毛细胞成功恢复其表达,ABR阈值恢复至接近野生型小鼠水平,听力恢复达7周以上,同时观察到内毛细胞带状突触形态部分改善,小鼠惊吓反应部分恢复。吴皓团队以AAV8为载体,通过后半规管注射,在5、8、20周龄的Vglut3基因敲除小鼠耳蜗内毛细胞中实现VGLUT3的外源性表达,听觉表型的恢复可持续12周以上,提示成年后基因治疗VGLUT3基因突变所致耳聋的有效性[34]。Mathiesen等[35]经枕大池向脑脊液中注射携带Vglut3基因的AAV,通过耳蜗导水管弥散进入内耳,挽救成年Vglut3基因敲除小鼠的听力表型,在大脑中检测到少量异位表达,肝脏中无异位表达,表明脑脊液递送是内耳基因治疗的可行递送途径。
2. OTOF基因:Otoferlin蛋白由OTOF基因编码,是耳蜗带状突触处的特异性钙离子结合蛋白,其作为钙浓度传感器发挥作用,驱动囊泡胞吐释放、融合以及带状突触活性区囊泡的补充[47, 48]。OTOF基因突变导致常染色体隐性耳聋DFNB9,患者常表现为语前聋,中度至重度听力损失,部分患者表现出温度敏感性[49, 50]。OTOF基因敲除小鼠听力损失严重,尽管存在正常的带状突触形态并能检测到Ca2+电流,但其带状突触的胞吐几乎完全被阻断[51],且无法维持带状突触活性区的囊泡补充速率[52]。
OTOF基因缺失早期不影响毛细胞的存活,为基因治疗提供足够的窗口期;治疗所面临的主要瓶颈是OTOF基因较大(6.0 kb),超过单个AAV的包装能力(4.7 kb),使得其难以通过单个AAV载体递送完成治疗。Reisinger等[36]首先尝试利用AAV递送较小的钙离子传感器Synaptotagmin1基因,但未能有效恢复小鼠带状突触的胞吐作用。Rankovic等[37]对耳蜗外植体和新生小鼠圆窗递送装载全长OTOF基因的过载AAV,可在约30%的内毛细胞恢复Otoferlin的特异性表达,实现OTOF基因敲除小鼠听力部分恢复。近来证实反式剪切策略可将较大的基因切割后分别包装到2个AAV,双病毒携带的质粒可在靶细胞翻译重组成完整蛋白[53]。Akil等[38]采用双AAV载体分别包装鼠源Otoferlin cDNA的5 ′片段部分以及3 ′片段部分,通过单次圆窗注射将双病毒重组载体递送至OTOF基因敲除小鼠的耳蜗中,形成重组cDNA编码序列,最终产生Otoferlin全长蛋白,新生小鼠注射4周后以及成年小鼠注射3周后均可测得其ABR阈值恢复至接近野生型水平。Al-Moyed等[39]也采用双AAV递送的形式,将OTOF基因递送至6~7日龄OTOF基因敲除小鼠的耳蜗中,高达50%的内毛细胞中均可表达Otoferlin全长蛋白,Otoferlin依赖的囊泡补充水平可达到野生型小鼠的35%~50%,听力得到部分恢复。
舒易来团队开发一种基于蛋白质反式剪接原理的双AAV介导的基因治疗系统,递送人源OTOF基因至OTOF基因敲除小鼠耳蜗中,注射侧听力可恢复到接近野生型水平并维持至少6个月[40],AAV1-hOTOF临床前研究表明其安全性良好[54],2022年复旦大学附属眼耳鼻喉科医院在国内率先开展OTOF基因治疗临床试验,最终招募6例OTOF基因突变所致常染色体隐性遗传严重至完全性听力损失患儿(中位年龄4.1岁),其中5例接受AAV1-hOTOF双病毒治疗的患儿在4~6周后均有听力获益,26周临床随访证实该病毒基因治疗安全有效[41]。
柴人杰团队采用双AAV-Anc80L65载体,有效恢复OTOF p.Q939X点突变小鼠听力,并在非人灵长类动物中证实其安全性。随后临床试验也取得成功:单侧单次圆窗给药的5岁患儿及双侧圆窗给药的8岁患儿在1个月后均有明显听力恢复,治疗后随访3个月未发现严重AAV-OTOF相关不良事件[42]。
总体而言,针对OTOF的双AAV递送基因过表达治疗研究已相对成熟,国内外正积极推动基于双AAV的多项耳聋基因疗法的临床试验研究,更大规模、更长随访时间的临床试验将评估耳聋基因治疗的长期有效性和安全性。
3. CaBP2基因:CaBP(calcium binding proteins)是与钙调蛋白相关的Ca2+结合蛋白家族,主要分布于中枢及感觉器官。CaBP2蛋白可对内毛细胞电压门控钙通道CaV1.3进行有效调节,控制突触前Ca2+内流,从而引起谷氨酸囊泡释放[55]。CaBP2的遗传缺陷导致非综合征型听力障碍DFNB93,为稳定的语前性听力损失,中度至重度[55, 56]。在小鼠体内敲除CaBP2会导致Ca2+通道的稳态失活,从而限制有效的突触囊泡传递[57],4周龄时小鼠ABR阈值显著升高,并进行性加重[58]。CaBP2敲除小鼠模型的毛细胞具有正常的突触发育情况及机械转导功能,为基因治疗提供足够的治疗窗口期[57, 58]。Oestreicher等[43]在CaBP2敲除小鼠上分别利用AAV1和AAV-PHP.eB两种病毒载体,将鼠源CaBP2基因递送至耳蜗,在5~8周龄时检测到听力改善,并恢复内毛细胞CaV1.3钙电流特性。
4. PJVK基因:PJVK基因与常染色体隐性遗传性耳聋DFNB59相关,其编码的Pejvakin蛋白表达于耳蜗Corti器、螺旋神经节细胞以及前三级听觉传入通路的神经元胞体,功能改变影响听觉传入通路[59]。PJVK基因突变患者听力学表型多变,Delmaghani等[59]在伊朗家系中发现PJVK p.R183W、p.T54I错义突变位点会导致非综合征型、语前性、重度到极重度的听力损失,听力学表型符合AN的诊断标准,而另一土耳其家系中发现的PJVK p.R183W错义突变与p.R167X无义突变[60]、p.Lys41GlufsX8移码突变[61]、p.R136X无义突变[62]患者的ABR和OAE均无法引出。Delmaghani等[44]发现Pejvakin蛋白与过氧化物酶体相关,在噪声刺激下诱导毛细胞内生成过氧化物酶体,在PJVK敲除小鼠中使用AAV8递送PJVK基因可部分恢复小鼠听力。
5. 综合征型听神经病相关基因:除了非综合征型耳聋,部分综合征型耳聋也有AN的表型。对于表现为AN的综合征型耳聋,耳聋的基因治疗尚无报道;但其他系统障碍的基因治疗已获成功,为基因治疗治愈综合征型AN提供新希望(表2)。
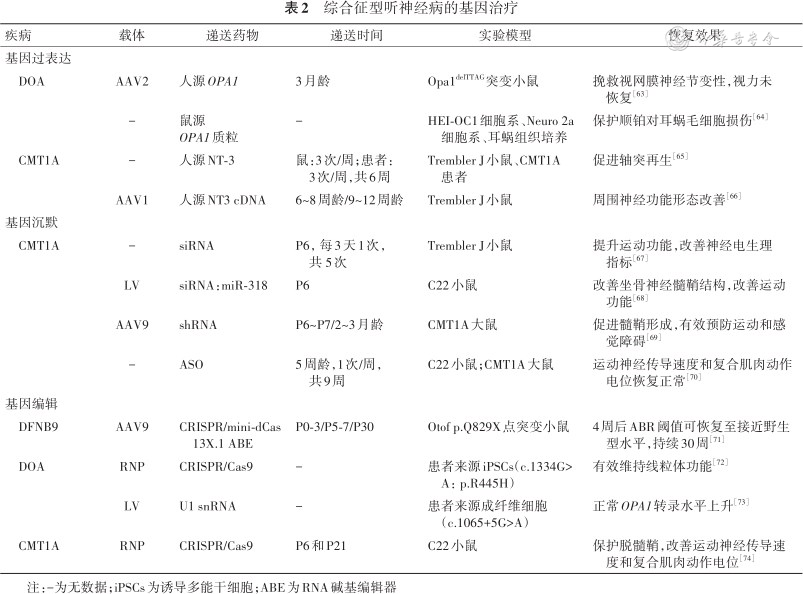
综合征型听神经病的基因治疗
综合征型听神经病的基因治疗
| 疾病 | 载体 | 递送药物 | 递送时间 | 实验模型 | 恢复效果 |
|---|---|---|---|---|---|
| 基因过表达 | |||||
| DOA | AAV2 | 人源OPA1 | 3月龄 | Opa1delTTAG突变小鼠 | 挽救视网膜神经节变性,视力未恢复[63] |
| - | 鼠源 OPA1质粒 | - | HEI-OC1细胞系、Neuro 2a细胞系、耳蜗组织培养 | 保护顺铂对耳蜗毛细胞损伤[64] | |
| CMT1A | - | 人源NT-3 | 鼠:3次/周;患者:3次/周,共6周 | Trembler J小鼠、CMT1A患者 | 促进轴突再生[65] |
| AAV1 | 人源NT3 cDNA | 6~8周龄/9~12周龄 | Trembler J小鼠 | 周围神经功能形态改善[66] | |
| 基因沉默 | |||||
| CMT1A | - | siRNA | P6,每3天1次,共 5次 | Trembler J小鼠 | 提升运动功能,改善神经电生理指标[67] |
| LV | siRNA:miR-318 | P6 | C22小鼠 | 改善坐骨神经髓鞘结构,改善运动功能[68] | |
| AAV9 | shRNA | P6~P7/2~3月龄 | CMT1A大鼠 | 促进髓鞘形成,有效预防运动和感觉障碍[69] | |
| - | ASO | 5周龄,1次/周,共9周 | C22小鼠;CMT1A大鼠 | 运动神经传导速度和复合肌肉动作电位恢复正常[70] | |
| 基因编辑 | |||||
| DFNB9 | AAV9 | CRISPR/mini-dCas13X.1 ABE | P0-3/P5-7/P30 | Otof p.Q829X点突变小鼠 | 4周后ABR阈值可恢复至接近野生型水平,持续30周[71] |
| DOA | RNP | CRISPR/Cas9 | - | 患者来源iPSCs(c.1334G>A:p.R445H) | 有效维持线粒体功能[72] |
| LV | U1 snRNA | - | 患者来源成纤维细胞(c.1065+5G>A) | 正常OPA1转录水平上升[73] | |
| CMT1A | RNP | CRISPR/Cas9 | P6和P21 | C22小鼠 | 保护脱髓鞘,改善运动神经传导速度和复合肌肉动作电位[74] |
注:-为无数据;iPSCs为诱导多能干细胞;ABE为RNA碱基编辑器
常染色体显性视神经萎缩(autosomal dominant optic atrophy,DOA)是最常见的遗传性视神经病变之一,多数病例由编码线粒体动力蛋白相关GTP酶的OPA1基因突变引起[75]。其特点是儿童期发病,视网膜神经节细胞的进行性丧失导致中心视力逐渐受损,最终出现视神经萎缩[76]。OPA1基因的错义突变所引起的显性负效应会导致综合征型常染色体显性视神经萎缩(DOA+),患者出现包括听力障碍、感觉运动神经病变、肌病和共济失调的表现[75]。DOA+相关听力障碍通常与OPA1基因14号外显子R445H错义突变相关,该突变可引起视神经和听神经的脱髓鞘以及突触丢失[75,77],听力受损患者的听力学检查结果符合听神经病诊断[78]。DOA+患者的耳聋往往随视力衰竭出现,高峰年龄段为20~30岁[75]。过表达OPA1是治疗DOA的可能方式,Sarzi等[63]向3月龄Opa1delTTG转基因小鼠的玻璃体内注射AAV2-OPA1,有效挽救视网膜神经节细胞的变性,尽管未取得明显视力功能恢复,该研究首次证明基因治疗DOA的可能性。Dong等[64]在内耳细胞系及培养的耳蜗组织上证实上调OPA1可以保护耳毒性药物诱导的耳蜗损伤,提示上调OPA1治疗DOA+相关听力障碍的可能性。
Charcot-Marie-Tooth(CMT)病是常染色体显性遗传性周围神经病变,主要分为2个亚型:1型脱髓鞘神经性病变(CMT1)和2型轴突神经性病变(CMT2)[79]。CMT1A患者典型临床表现包括远端肌无力、感觉丧失以及运动和感觉神经传导速度缓慢[79],部分患者听神经亦发生脱髓鞘病变,受累患者在嘈杂环境下的言语感知能力明显下降,时间和频谱分辨率也下降[80]。神经营养因子3(neurotrophin-3,NT-3)是施万细胞自分泌回路的重要组成部分,与神经元成熟、存活以及促轴突再生功能密切相关,可改善CMT1A小鼠模型中的神经再生和髓鞘形成能力[81]。Sahenk等[65]向CMT1A患者皮下注射NT-3,观察到有髓纤维数量的增多以及神经生理学功能的改善;该团队将AAV1包装的NT-3 cDNA注射至Pmp22点突变Trembler J(Tr-J)小鼠肌肉中,最终在血液内检测到NT-3水平升高且能改善小鼠周围神经的形态及功能[66]。NT-3对AN的改善作用尚无报道。
基因沉默是利用基因调控技术抑制某些基因的表达,对于显性突变可通过减少致病基因的表达来实现疾病的治疗。在转录水平上,可通过CRISPRi(CRISPR interference)系统和CRISPR/dCas9系统进行表观遗传修饰来实现转录抑制,在转录后水平可利用反义寡聚核苷酸(antisense oligonucleotides,ASO)和RNA干扰(RNA interference,RNAi)策略。在脊髓性肌萎缩症、转甲状腺素蛋白介导(ATTR)淀粉样变性心肌病领域已有基于基因沉默疗法的获批药物[82]。
CMT1A型脱髓鞘神经病由外周髓鞘蛋白PMP22基因重复拷贝导致[83],其过表达引起脱髓鞘神经病变[84, 85],亦累及听神经[80],因此运用基因治疗手段下调PMP22基因的过表达是治疗CMT1A的合理策略之一。Lee等[67]设计19种不同的Pmp22等位基因特异性siRNA,在Tr-J小鼠模型上进行验证,筛选出的siRNA能特异性降低Pmp22基因的表达水平,增加小鼠施万细胞髓鞘蛋白表达,提升小鼠的运动功能;随后该团队利用miR-318有效控制Pmp22基因拷贝数的异常,实现形态以及功能的恢复[68]。Gautier等[69]采用AAV9载体,在CMT1A大鼠坐骨神经内注射表达靶向Pmp22基因的shRNA,使得PMP22蛋白水平正常化,髓鞘形成增加,运动和感觉障碍的恢复可持续12个月。ASO也可有效降低Pmp22 mRNA水平,C22小鼠及CMT1A大鼠模型皮下注射ASO后,其运动神经传导速度和复合肌肉动作电位均可恢复至接近野生型动物水平[70]。
无论是siRNA还是ASO,均需要反复给药以抑制基因表达,后续研究还需明确其最佳的给药途径和频率以及长期治疗的疗效问题。
基因编辑技术通过删除、插入或替换基因组的某个片段或特定碱基使基因组发生特定变化,以干扰致病基因或纠正致病碱基突变。随着CRISPR/Cas9基因编辑效率的提升,基因编辑技术深刻改变了生物医药行业,目前已开展多项临床试验,2023年底基于CRISPR/Cas9的体外造血干细胞基因编辑疗法Casgevy作为第一款基因编辑药物已受获批上市[86]。CRISPR/Cas13系统靶向的RNA编辑不改变基因组,其介导的基因组编辑具有可逆性,理论上有更好的安全性。
OTOF突变在我国婴幼儿AN患病人群中占41.2%[50],随着小型、高效的基因编辑工具不断迭代,高效在体基因编辑修正OTOF点突变成为可能。吴皓团队采用单个AAV为载体携带小尺寸CRISPR/Cas13 RNA单碱基编辑工具(emxABE),精准修复人源化Otof p.Q829X点突变小鼠模型中mRNA中突变位点,新生及成年小鼠治疗后听力接近野生型水平,证实单AAV递送可以治疗OTOF遗传性耳聋,为遗传性耳聋精准基因治疗策略的开发开辟出一片新领域[71]。
OPA1的c.1334G>A(p.R445H)突变为DOA+致病突变之一,针对该突变体,Sladen等[72]采用CRISPR/Cas9基因编辑工具处理患者来源的多能干细胞,对该处突变进行修正达到57%的基因编辑效率,并有效维持线粒体稳态,恢复基础呼吸以及ATP生成水平,同时,对OPA1突变的编辑能够重建野生型mtDNA的水平,降低线粒体对凋亡的易感性,为后续治疗动物疾病模型提供了基础。
单倍体剂量不足是DOA的主要遗传致病机制,其中近30%的OPA1相关病例出现剪接位点突变。RNA剪接是成熟mRNA形成前的重要一步,由剪接体复合物完成,5个不同的小核RNA(small nuclear RNA,snRNA)亚基以及一系列相关的蛋白辅助因子组成剪接体U1、U2、U4、U5、U6,共同在细胞核内完成剪接[87]。OPA1基因突变位点c.1065+5G>A可导致OPA1基因10号外显子跳跃,使得OPA1蛋白表达减少约50%,该突变可能阻碍了启动剪接过程所需的重要剪接体U1对剪接位点的识别。Jüschke等[73]对剪接体U1的5′ 部分进行改造,使其能通过碱基互补配对的方式重新定位到OPA1基因10号外显子或10号内含子的不同位置,在患者来源的成纤维细胞中,将U1剪接因子结合到10号内含子的+18位点可以有效回避剪接突变导致的10号外显子的跳跃,从而增加正常转录本表达水平。该研究提示靶向剪接突变位点进行改造可以作为治疗DOA的新思路。
针对CMT1A型脱髓鞘神经病重复拷贝导致的脱髓鞘病变,Lee等[74]运用CRISPR/Cas9工具靶向Pmp22 P1启动子的TATA-box元件,实现Pmp22 mRNA水平下调,改善神经病理症状,能有效防止PMP22表达过低导致遗传性压力易感性周围神经病。
随着OTOF耳聋患者经基因治疗后听力的恢复,证实耳聋基因治疗的临床有效性,为耳聋的治疗提供了新的治疗方案。但目前耳聋基因治疗临床转化主要集中于OTOF基因,尚无其他遗传性耳聋基因治疗临床转化的报道。究其原因,一方面是OTOF主要表达于内毛细胞,目前AAV对内毛细胞靶向性非常好,基本能实现全部内毛细胞的转染。另外,OTOF突变导致的听力障碍不影响毛细胞的存活,为基因治疗提供了足够的窗口期。AN的基因治疗,乃至耳聋的基因治疗,目前最大的瓶颈在于是否有足够治疗窗口。
基因治疗可以实现遗传性疾病的根治,耳聋基因治疗可以恢复患者的自然聆听,也可以避免人工听觉植入体的弊端,在遗传性耳聋治疗领域有很好的应用前景。由于部分AN人工耳蜗植入效果不佳,基因治疗有更广阔的应用价值。基因过表达策略已经证实可以治疗遗传性耳聋,而更精准的基因编辑工具将能实现AN致病突变的在体修改,最终实现一次治疗、终生获益。
孙怡琳, 金晨曦, 冯宝怡, 等. 听神经病的基因治疗现状[J]. 中华耳鼻咽喉头颈外科杂志, 2024, 59(5): 510-518. DOI: 10.3760/cma.j.cn115330-20231029-00177.
所有作者声明无利益冲突
1.以下关于听神经病的说法错误的是?(单选)
A.听性脑干反应无法引出或严重异常,耳声发射可引出
B.声导抗表现为鼓室图正常,镫骨肌反射消失或阈值升高
C.患者言语识别能力受损程度大于其听力损失程度
D.各类型听神经病患者均可通过人工耳蜗植入获得良好治疗效果
2.以下哪一项是非综合征型听神经病相关基因?(单选)
A.OPA1
B.MPZ
C.DIAPH3
D.PMP22
3.耳聋基因治疗推进的基础在于是否有足够治疗窗口,以下遗传性耳聋类型中相对具有足够基因治疗窗口期的是?(多选)
A.DFNA25
B.DFNB9
C.DFNB93
D.DFNB59
4.针对OTOF相关遗传性耳聋的基因治疗研究已相对成熟并逐步迈入临床转化阶段,在现有研究中以下可获得疗效的基因治疗模式是?(多选)
A.单AAV包载Synaptotagmin1基因
B.单AAV包载全长Otof
C.双AAV包载Otof重组片段cDNA
D.单AAV包载RNA单碱基编辑器
5.综合征型常染色体显性视神经萎缩(DOA+)由编码线粒体动力蛋白相关GTP酶的OPA1基因突变引起,其最常见的眼外体征是?(单选)
A.听力障碍
B.感觉和/或运动神经病变
C.肌病
D.共济失调继续教育园地参考答案见本期第446页

























