
原发性皮肤T细胞淋巴瘤(PCTCL)是一组原发于皮肤的罕见非霍奇金淋巴瘤,最常见亚型为蕈样肉芽肿,其次为原发性皮肤CD30阳性T细胞淋巴组织增生性疾病。虽然2022年第5版世界卫生组织造血淋巴肿瘤分类已将Sézary综合征从PCTCL中划除,但基于其与蕈样肉芽肿的密切相关性,依然有必要一起讨论。PCTCL的早期确诊和及时治疗十分重要,专家组成员结合PCTCL的诊治现状及国内外相关研究数据,制定了该共识,旨在为临床医生提供参考。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
原发性皮肤T细胞淋巴瘤(PCTCL)是一组原发于皮肤的非霍奇金淋巴瘤,占原发性皮肤淋巴瘤的75%~80%[1]。我国皮肤T细胞淋巴瘤(CTCL)发病率预估为6.9/100万[2]。PCTCL最常见的亚型为蕈样肉芽肿(MF),其次为原发性皮肤CD30阳性T细胞淋巴组织增生性疾病(PCTLD),在我国分别约占PCTCL的65%、13%[3]。PCTCL的病因暂不明确,发病可能与DNA损伤、表观遗传调控、程序性细胞死亡、T细胞受体(TCR)信号转导通路异常等有关[4,5]。
PCTCL的确诊主要基于患者临床表现和组织病理学结果的相关性。
PCTCL的疾病亚型、分期和临床表现均具有多样性,大多数(约90%)PCTCL患者可出现瘙痒[6]。
对于最常见的MF,经典型MF的早期表现为斑片、斑块,进展期可出现肿瘤或红皮病,也可能出现皮肤外受累(如区域淋巴结肿大)[4,6,7,8,9]。斑片期MF的皮损主要为大小不等、形态各异的淡红色至暗红色斑片或斑块,大多数直径>5 cm,表面有少量细碎鳞屑,未经治疗的皮损可缓慢增大或部分消退,并伴有不同程度的瘙痒[4,6,7,8,9]。斑块期MF表现为大小不等的暗红色、棕红色浸润性斑块,伴有不同程度的鳞屑。肿瘤期MF表现为斑片、斑块和肿瘤混合出现,肿瘤大小不一,可向皮肤表面隆起或如蕈样增生,常见溃疡,可伴有剧烈疼痛[4,6,7,8,9]。红皮病型MF/Sézary综合征(SS)以红皮病(红斑累及≥80%皮肤表面)为主要特征,还包括足底和手掌角化病、指甲营养不良/外翻、弥漫性脱发,并且淋巴结肿大常见,因此体格检查时建议进行全身浅表淋巴结触诊[4,6]。
大多数原发性皮肤间变性大细胞淋巴瘤(pcALCL)患者表现为孤立或聚集性结节,生长数周至数月,这些结节大部分会随着时间的推移而破溃;约20%的病例为多灶性疾病,约10%的病例会发生皮外病变,通常累及区域淋巴结[6]。淋巴瘤样丘疹病(LyP)表现为多发性皮肤丘疹、结节伴部分坏死,可出现自发消退和较为频繁的复发[6]。侵袭性亚型多表现为皮肤肿块,并可伴有全身症状,还有部分惰性肿瘤可表现为皮肤孤立性结节或水疱-痘疮样改变[8,9]。
对于皮肤病变提示PCTCL的患者,需进行皮肤活组织检查以验证淋巴瘤的诊断,病理检测技术包括组织形态学分析、免疫组织化学(IHC)、流式细胞术、TCR基因重排分析,并视情况对淋巴结或可疑的皮外病灶进行活组织检查[1]。建议活组织检查的取样体积为1 cm3,取样位置选择病变处和皮肤病变与正常组织的交界处。应由具有CTCL诊断专业知识的病理医生对充足标本组织和(或)有代表性病变组织的石蜡切片进行检查[6]。为避免药物因素可能造成的干扰,在活组织检查前应暂停局部治疗2周以上[7]。如果活组织检查结果无法确诊和(或)与临床表现不一致,需在接下来的几个月内再次进行活组织检查[6,7]。
对于MF/SS患者,建议对异常的肿大淋巴结或可疑的皮外部位进行活组织检查;对于pcALCL患者,当皮肤活组织检查无法确诊时可对肿大淋巴结或皮外部位进行活组织检查[6]。选择切除或切取活组织检查,仅细针吸取活组织检查不足以初步诊断淋巴瘤[6]。建议IHC常规检查指标包括CD20、CD3、CD4、CD5、CD7、CD8、CD30、CD56、TIA1、EBER、Ki-67,拓展检查指标包括TCRα、TCRβ、CD25、CCR4、PD-1、CXCL13、ICOS、ALK[1]。MF大细胞转化可表达CD30。
PCTCL的诊断所需要的辅助检查项目包括完善病史采集和体格检查、实验室检查,视情况进行影像学检查(表1)[1,6,7,10]。

| 检查类型 | 建议项目 |
|---|---|
| 病史采集和体格检查 | B症状 |
| 皮肤检查a | |
| 浅表淋巴结、腹部触诊 | |
| 体能状态检查 | |
| 实验室检查 | 全血细胞计数 |
| 肝肾功能、电解质检查 | |
| 含乳酸脱氢酶的血液生化检查b | |
| 妊娠检查(育龄妇女) | |
| 外周血Sézary细胞流式细胞术检查(包括CD4+ CD7-或CD4+ CD26-)c | |
| 外周血涂片(红皮病) | |
| 影像学检查 | 胸部、腹部、盆腔CT检查或全身PET-CT检查d |
注:a包括病变累及占体表面积百分比(推荐使用改良的严重程度加权评估工具进行计算[10])及皮肤病变(红斑、肿物、斑块等);b蕈样肉芽肿/Sézary综合征还需检测β2-微球蛋白;c针对≥T2期蕈样肉芽肿/Sézary综合征患者,进行该检查以评估和定量具有异常表型的扩增T细胞群;dT3~T4期蕈样肉芽肿/Sézary综合征患者进行全身疾病评估时,PET-CT扫描范围需包括手臂和腿,如未做过全身PET-CT,则进行颈部增强CT,而淋巴瘤样丘疹病只有当怀疑相关淋巴瘤累及全身时才进行影像学检查
PCTCL分类主要参考2022年第5版世界卫生组织(WHO)造血淋巴肿瘤分类[11],包括MF(包括MF变异型,如亲毛囊性MF、Paget样网状细胞增生症、肉芽肿性皮肤松弛症)、原发性皮肤CD30阳性T细胞淋巴组织增生性疾病(包括LyP、pcALCL)、皮下脂膜炎样T细胞淋巴瘤、原发性皮肤γ/δT细胞淋巴瘤、原发性皮肤CD8阳性侵袭性嗜表皮性细胞毒性T细胞淋巴瘤、原发性皮肤CD4阳性小或中T细胞淋巴组织增生性疾病、原发性皮肤肢端CD8阳性淋巴组织增生性疾病、原发性皮肤外周T细胞淋巴瘤,非特指型(PTCL-NOS)。因MF和SS分别起源于皮肤组织常驻记忆T细胞和胸腺记忆T细胞,虽然二者密切相关,但有观点认为SS不同于MF[6]。在2022年第5版WHO造血淋巴肿瘤分类中SS被归入成熟T细胞及自然杀伤(NK)细胞白血病类别[11]。
MF/SS的分期通常采用TNMB系统,该系统包括对皮肤(T)、淋巴结(N)、内脏(M)和血液(B)受累的评估,其中淋巴结的组织病理学分期主要应用美国国家癌症研究所(NCI)-VA标准/Dutch标准[12] 。MF/SS的TNMB分期及临床分期具体方法见表2,表3,表4,表5,表6。

蕈样肉芽肿/Sézary综合征的T分期方法[12]
蕈样肉芽肿/Sézary综合征的T分期方法[12]
| T分期 | 定义 |
|---|---|
| T0 | 临床无皮肤病变 |
| T1 | 局限性斑块、丘疹和(或)皮肤受累范围<10%体表面积 |
| T1a | 仅有斑块 |
| T1b | 丘疹或丘疹与斑块 |
| T2 | 斑块、丘疹和(或)皮肤受累范围≥10%体表面积 |
| T2a | 仅有斑块 |
| T2b | 丘疹或丘疹与斑块 |
| T3 | 任何肿块 |
| T4 | 皮肤红斑≥80%体表面积 |
注:T分期为皮肤受累程度分期;斑块为任何大小皮损,无隆起或硬结,注意皮下色素沉着、脱屑、结节和(或)皮肤异色;丘疹为任何大小皮损,隆起或硬结,注意脱屑、结节和(或)皮肤异色、溃疡;肿块为一个以上直径>1 cm的实性或结节性病变,有向深部生长的迹象,注意病变总数、总体积、最大病变面积和病变累及部位

蕈样肉芽肿/Sézary综合征的N分期方法[12]
蕈样肉芽肿/Sézary综合征的N分期方法[12]
| N分期 | 定义 |
|---|---|
| N0 | 无临床异常的外周淋巴结,无需活组织检查 |
| N1 | 临床异常外周淋巴结,组织病理Dutch分级1或NCI LN0~2 |
| N1a | 无法确定单克隆性 |
| N1b | 克隆性与皮肤病变一致 |
| N2 | 临床异常外周淋巴结,组织病理Dutch分级2或NCI LN3 |
| N2a | 无法确定单克隆性 |
| N2b | 克隆性与皮肤病变一致 |
| N3 | 临床异常外周淋巴结,组织病理Dutch分级3~4或NCI LN4 |
| N3a | 无法确定单克隆性 |
| N3b | 克隆性与皮肤病变一致 |
| NX | 临床异常外周淋巴结,无组织病理证据 |
注:N分期为淋巴结受累程度分期;NCI为美国国家癌症研究所

蕈样肉芽肿/Sézary综合征的M分期方法[12]
蕈样肉芽肿/Sézary综合征的M分期方法[12]
| M分期 | 定义 |
|---|---|
| M0 | 无脏器受累 |
| M1 | 脏器受累(由组织病理确诊,且器官受累必须是特异性的) |
| M1a | 仅骨髓受累 |
| M1b | 非骨髓脏器受累 |
| MX | 脏器病变(无病理证实) |
注:M分期为内脏受累程度分期
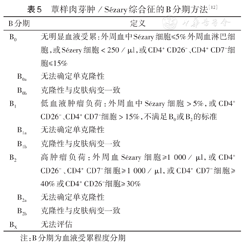
蕈样肉芽肿/Sézary综合征的B分期方法[12]
蕈样肉芽肿/Sézary综合征的B分期方法[12]
| B分期 | 定义 |
|---|---|
| B0 | 无明显血液受累:外周血中Sézary细胞≤5%外周血淋巴细胞,或Sézery细胞<250/μl,或CD4+ CD26-、CD4+ CD7-细胞≤15% |
| B0a | 无法确定单克隆性 |
| B0b | 克隆性与皮肤病变一致 |
| B1 | 低血液肿瘤负荷:外周血中Sézary细胞>5%,或CD4+ CD26-、CD4+ CD7-细胞>15%,不满足B0或B2的标准 |
| B1a | 无法确定单克隆性 |
| B1b | 克隆性与皮肤病变一致 |
| B2 | 高肿瘤负荷:外周血Sézary细胞≥1 000/μl,或CD4+ CD26-、CD4+ CD7-细胞≥1 000/μl,或CD4+ CD7-细胞≥40%或CD4+ CD26-细胞≥30% |
| B2a | 无法确定单克隆性 |
| B2b | 克隆性与皮肤病变一致 |
| BX | 无法评估 |
注:B分期为血液受累程度分期

蕈样肉芽肿/Sézary综合征的临床分期方法[12]
蕈样肉芽肿/Sézary综合征的临床分期方法[12]
| 临床分期 | T | N | M | B |
|---|---|---|---|---|
| ⅠA(局限性皮肤受累) | T1 | N0 | M0 | B0/B1 |
| ⅠB(仅皮肤病变) | T2 | N0 | M0 | B0/B1 |
| ⅡA | T1/T2 | N1/N2 | M0 | B0/B1 |
| ⅡB(肿瘤期疾病) | T3 | N0/N1/N2 | M0 | B0/B1 |
| ⅢA(红皮病) | T4 | N0/N1/N2 | M0 | B0 |
| ⅢB(红皮病) | T4 | N0/N1/N2 | M0 | B1 |
| ⅣA1(Sézary综合征) | T1/T2/T3/T4 | N0/N1/N2 | M0 | B2 |
| ⅣA2(Sézary综合征或非Sézary综合征) | T1/T2/T3/T4 | N3 | M0 | B0/B1/B2 |
| ⅣB(脏器疾病) | T1/T2/T3/T4 | N0/N1/N2/N3 | M1 | B0/B1/B1 |
| 大细胞转化 | ||||
对于除MF/SS外包含PCTLD的PCTCL,可应用TNM分期系统进行分期[13],具体分期方法见表7,表8,表9。

除蕈样肉芽肿/Sézary综合征外的原发性皮肤T细胞淋巴瘤的T分期方法[13]
除蕈样肉芽肿/Sézary综合征外的原发性皮肤T细胞淋巴瘤的T分期方法[13]
| T分期 | 定义 |
|---|---|
| T1 | 孤立皮肤病变 |
| T1a | 单一孤立病变且直径<5 cm |
| T1b | 单一孤立病变且直径≥5 cm |
| T2 | 区域皮肤受累a:多个病变局限于1个区域或2个连续区域 |
| T2a | 所有病变总体直径<15 cm |
| T2b | 所有病变总体直径≥15 cm但<30 cm |
| T2c | 所有病变总体直径≥30 cm |
| T3 | 广泛皮肤累及 |
| T3a | 多处病变但局限于2个连续区域内 |
| T3b | 多处病变累及≥3个区域 |
注:a区域皮肤受累及对应体表面积


CTCL的预后因素包括是否存在皮肤外受累、皮肤受累的体表面积、皮肤病变类型等[14]。MF的总体预后良好,特异性5年总生存(OS)率为88%,但进展期MF预后明显较差(特异性5年OS率:ⅠA期95.7%,Ⅳ期23.6%),预后不良因素还包括年龄>60岁、乳酸脱氢酶水平升高、伴大细胞转化;SS预后较差,5年OS率为10%~50%[1,15,16];血液受累也是MF/SS重要的预后因素,与OS差相关(P<0.001)[17]。
PCTLD总体预后良好,其中LyP的10年特异性OS率接近100%,但15%~50%会发生继发性恶性肿瘤,主要为二次CTCL,相关临床因素未知;pcALCL的5年特异性OS率达95%,预后还与受累部位有关,腿部受累与广泛肢体疾病(ELD)是预后不良因素[9,15,18]。
MF的治疗目标是控制病情、延缓复发和进展,延长生命,通常根据患者的症状、给药途径、不良反应和总体治疗目标给予个体化治疗[6,19]。MF及其变异型应按分期进行治疗。早期MF(ⅠA、ⅠB和ⅡA期)以皮肤定向治疗(SDT)为主,包括外用氮芥类药物、光疗(如窄谱中波紫外线)、外用糖皮质激素(GCS)、局部放疗[20]。外用GCS单药治疗可带来约33%的完全缓解(CR)率,但不建议长期使用超强效GCS(如丙酸氯倍他索),推荐使用中强效GCS,待皮疹消退后停用,无须长期维持[7,19]。对于皮肤广泛受累、皮肤疾病负担较重、主要表现为斑块、血液受累和(或)对SDT反应不佳的患者,应考虑系统治疗(单药或联合治疗)[6]。进展期MF(ⅡB~Ⅳ期)以系统治疗为主,常与长波紫外线1或其他疗法(如SDT中的GCS、氮芥类药物、全身电子束治疗)联合应用[20]。光疗和全身电子束治疗可能与红皮病患者的不良反应增加有关,可以考虑调整剂量或治疗时间[6]。多药化疗方案通常用于复发、难治或皮外受累的患者,大多数患者在接受多药化疗前接受多种单药系统治疗[6]。对于初始治疗获缓解和(或)具有临床获益的患者,应考虑维持治疗以优化缓解持续时间。对于影响患者生命质量的伴随症状,例如瘙痒(在进展期MF中更严重),可考虑GCS局部用药、润肤剂等[20]。治疗过程中应注意采取基于分期的规范治疗。MF的治疗流程见图1。
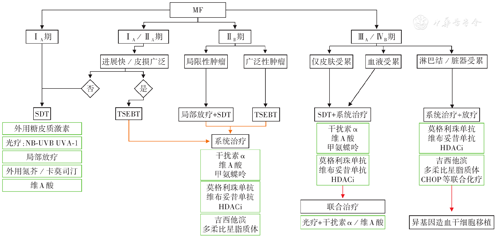
注: 为该疗法包含的具体药物及具体治疗方案;
为该疗法包含的具体药物及具体治疗方案; 所指方格表示一线治疗效果不佳时可选择的二线治疗方案;
所指方格表示一线治疗效果不佳时可选择的二线治疗方案; 所指方格表示可联合应用的疗法;MF为蕈样肉芽肿;SDT为皮肤定向治疗;NB-UVB为窄谱中波紫外线;UVA-1为长波紫外线1;TSEBT为全身电子束治疗;HDACi为组蛋白去乙酰化酶抑制剂;CHOP为环磷酰胺、多柔比星、长春新碱、泼尼松
所指方格表示可联合应用的疗法;MF为蕈样肉芽肿;SDT为皮肤定向治疗;NB-UVB为窄谱中波紫外线;UVA-1为长波紫外线1;TSEBT为全身电子束治疗;HDACi为组蛋白去乙酰化酶抑制剂;CHOP为环磷酰胺、多柔比星、长春新碱、泼尼松
由于感染并发症在MF患者中常见,尤其是细菌感染和疱疹病毒感染,可通过常规使用皮肤保湿剂和(或)润肤剂保护皮肤屏障,避免使用中心静脉导管等,以降低皮肤感染风险[6]。对于继发皮肤感染风险高的患者(例如红皮病),应密切监测并考虑预防性使用抗菌药物或抗病毒药物,必要时使用抗菌药物行经验性治疗[6]。
LyP的首要治疗策略是观察病情变化,可基于疾病严重程度和患者意愿选择治疗方案,无症状患者优先随访观察,有症状、进展/转化证据的患者需要干预[21] 。(1)SDT:局部GCS或光疗是局部病变或广泛病变患者的初始治疗选择,窄谱中波紫外线通常优于光化学疗法,停止治疗后容易复发,局部放疗、低剂量全身电子束治疗也被用于难治、病变广泛的患者;(2)系统治疗适用于病变广泛的患者,甲氨蝶呤被广泛用于LyP的系统治疗,可考虑在CR后3~6个月尝试逐步戒断[6,22]。目前的治疗方案可有效地控制疾病但不能防止疾病进展。对于局部或轻度病变患者,在避免过度治疗疾病的同时仔细观察是可行的选择。
pcALCL应根据皮外受累情况制订治疗方案,对于单发或局限性病灶可选择局部放疗、手术切除联合或不联合局部放疗;对于广泛性病灶或复发难治疾病,可考虑维布妥昔单抗,或低剂量甲氨蝶呤、维A酸、普拉曲沙、干扰素等联合或不联合SDT,对于局部淋巴结受累的患者也可以考虑环磷酰胺、多柔比星、长春新碱、泼尼松(CHOP方案)多药化疗[6,23]。
其他PCTCL可根据疾病的侵袭性进行治疗选择,对于原发性皮肤γ/δT细胞淋巴瘤和原发性皮肤CD8阳性侵袭性嗜表皮性细胞毒性T细胞淋巴瘤这类侵袭性亚型,建议根据欧洲肿瘤内科学会(ESMO)PTCL-NOS指南进行治疗[24,25]。对于原发性皮肤CD4阳性小或中T细胞淋巴组织增生性疾病和原发性皮肤肢端CD8阳性淋巴组织增生性疾病等较惰性的亚型,患者预后良好,通常表现为单一皮肤病变,可考虑局部放疗或手术切除治疗[24]。
尽管SS在2022年第5版WHO造血淋巴肿瘤分类中已不属于PCTCL,但基于其与MF密切相关,目前国内外指南的治疗推荐中仍未将SS与MF分开。SS的初始治疗应根据肿瘤负荷分层,低中负荷(Sézery细胞<5 000/mm3)患者可考虑系统治疗±SDT,高负荷(Sézery细胞>5 000/mm3)患者可考虑组蛋白去乙酰化酶抑制剂(HDACi)±SDT或系统治疗、莫格利珠单抗±SDT[1]。系统治疗包括干扰素α、甲氨蝶呤、莫格利珠单抗、HDACi,SDT包括光疗、局部GCS、氮芥类药物、全身电子束治疗(12~36 Gy)[1]。对于初始治疗获缓解和(或)具有临床获益的患者,应考虑维持治疗以优化缓解持续时间[6]。SS的治疗流程见图2。
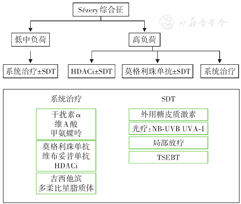
注: 为该疗法包含的具体药物及具体治疗方案;HDACi为组蛋白去乙酰化酶抑制剂;SDT为皮肤定向治疗;NB-UVB为窄谱中波紫外线;UVA-1为长波紫外线1;TSEBT为全身电子束治疗
为该疗法包含的具体药物及具体治疗方案;HDACi为组蛋白去乙酰化酶抑制剂;SDT为皮肤定向治疗;NB-UVB为窄谱中波紫外线;UVA-1为长波紫外线1;TSEBT为全身电子束治疗
免疫治疗药物是PCTCL系统治疗的重要组成部分,主要包括莫格利珠单抗、维布妥昔单抗、伏立诺他、罗米地辛、西达本胺等。由于SDT难治的早期和进展期MF/SS患者的系统治疗缓解率低且持续时间通常很短,至下次治疗时间(TTNT)可作为反映治疗获益的指标[20]。
CCR4单抗莫格利珠单抗在我国获批用于既往接受过系统性治疗的复发或难治SS或Ⅲ~Ⅳ期MF成年患者。全球Ⅲ期MAVORIC研究纳入既往接受过治疗的CTCL患者372例,中位随访17个月,莫格利珠单抗对比伏立诺他可延长无进展生存(PFS)时间(中位PFS时间:7.7个月比3.1个月)和TTNT(11个月比3.5个月),血液累及的患者具有更好疗效(TTNT:B1期12.6个月比3.1个月;B2期13.1个月比3.5个月;中位PFS时间:B1期8.63个月比2.53个月;B2期11.17个月比3.30个月),还可带来更高的血液学缓解率,并可有效缓解皮肤症状、降低皮肤肿瘤负荷[血液客观缓解率(ORR):68%比19%;皮肤ORR:42%比16%],明显改善瘙痒、疼痛症状(P<0.05)[26,27,28]。莫格利珠单抗的具体用药参考其说明书[29]。
靶向CD30的抗体偶联药物维布妥昔单抗在我国批准用于CD30阳性淋巴瘤成年患者,包括既往接受过系统性治疗的pcALCL和MF,主要基于全球Ⅲ期ALCANZA研究结果(纳入CD30阳性pcALCL和MF患者131例,不含SS),中位随访45.9个月,对比医生选择治疗组(口服甲氨蝶呤或贝沙罗汀),维布妥昔单抗组持续至少4个月的ORR(ORR4)显著更优(ORR4:54.7%比12.5%,P<0.001);中位随访36.8个月,维布妥昔单抗组中位PFS时间(16.7个月比3.5个月,P<0.001)以及TTNT(14.2个月比5.6个月,P<0.001)明显延长[30]。在Skindex-29症状负担减轻程度方面,与医生选择治疗组相比,维布妥昔单抗组更优(-27.9比-8.62,P<0.001)[31]。
在HDACi中,伏立诺他、罗米地辛治疗CTCL的ORR为30%~40%,但目前尚未在我国获批;西达本胺在我国获批用于外周T细胞淋巴瘤的治疗,在CTCL中也有应用[6]。
此外,一些正在探索的免疫治疗药物包括CD52单抗alemtuzumab已被美国食品药品管理局(FDA)批准用于治疗伴或不伴血液受累的红皮病型MF(Ⅲ~ⅣA期)患者,既往研究显示进展期MF/SS患者的ORR为51%~55%,但在我国尚未获批[7]。免疫检查点抑制剂如程序性死亡受体1单抗帕博利珠单抗在MF/SS中的ORR为38%,中位随访58周时中位缓解持续时间(DOR)未达到[6]。
根据不同治疗药物,参照常见不良反应术语评定标准(CTCAE)5.0版对不良反应的严重程度进行分级管理。
莫格利珠单抗最常见不良反应是输液反应和药物性皮炎(药疹),大部分为1~2级。在整个治疗过程中需监测患者是否出现皮疹,建议行皮肤活组织检查来区分疾病进展与药疹。对于药疹的处理措施包括局部外用皮质类固醇(1级皮疹)、中断(2~3级皮疹)或永久终止(4级皮疹)莫格利珠单抗治疗[29]。输液反应大多数(约90%)发生在首次输液期间或之后不久,可考虑在首次输注前给予预治疗(如给予苯海拉明和对乙酰氨基酚),但尚未确定预治疗是否降低了这些反应的发生风险或严重程度。
维布妥昔单抗最常见的不良反应是周围神经病变(PN),也是导致治疗终止的最常见不良反应。PN在CTCL中的发生率为67%,大部分是1~2级,多数患者在完成或停止治疗后,PN会消失或改善。如治疗期间出现周围感觉或运动神经病变或PN加重,可能需要推迟给药、调整维布妥昔单抗剂量或终止治疗[32]。
西达本胺最常见不良反应是血液学不良反应和胃肠道反应。血液学不良反应多发生在首次服药后6周内,可给予对症支持治疗,减量或暂停用药可有效缓解;胃肠道反应多为1~2级,建议继续用药,维持原剂量并密切进行临床观察随访和实验室检查,如发生3级以上不良反应则暂停用药,予对症支持治疗或紧急治疗,2周内不良反应≤1级后恢复用药[33]。
MF/SS的疗效评估目前主要参考国际皮肤淋巴瘤学会(ISCL)、美国皮肤淋巴瘤协会(USCLC)及欧洲肿瘤研究与治疗组织(EORTC)共同提出的MF/SS缓解标准[10]。PCTLD的疗效评估目前主要参考ISCL、USCLC、EORTC共同提出的PCTLD缓解标准[23]。
常见PCTCL的随访应根据临床情况制订个体化的随访计划,随访时应重点关注病史和体格检查[24]。MF患者达到CR或部分缓解(PR)后,可每3~6个月随访1次,随访包括皮损变化、浅表淋巴结大小(B型超声)、血液检查(包括血常规、乳酸脱氢酶、β2-微球蛋白、外周血异型细胞,必要时行外周血流式细胞术检测)、新发皮疹组织病理检查、肿大淋巴结组织病理检查,必要时行CT或PET-CT检查和骨髓检查[18]。在PCTLD中,由于LyP患者发生二次淋巴恶性肿瘤的风险可达15.5%(即使是初始治疗有效的患者),需终身随访,包括全面皮肤检查;pcALCL随访期间可能出现MF,因此随访期间应持续进行全面的皮肤检查[6,34]。
由于PCTCL患者的生命质量和心理健康往往受疾病影响严重,除积极治疗以提高生命质量外,还可开展宣教活动,帮助患者正确认识疾病。
PCTCL是一种罕见、病因不明且具有复杂免疫学背景的异质性淋巴瘤。PCTCL的异质性增加了其常见标志物和(或)治疗靶点检测的难度,未来新技术与人工智能的结合有望助力PCTCL的个体化治疗[4]。目前新型靶向免疫治疗药物正在探索中,期待能够提供更多有效且不良反应小的药物用于治疗PCTCL。
执笔 马军、朱军、赵东陆
审阅 沈志祥
专家组成员(按姓氏汉语拼音字母排序) 白鸥(吉林大学白求恩第一医院)、曹军宁(复旦大学附属肿瘤医院)、陈浩(中国医学科学院皮肤病医院)、侯健(上海交通大学医学院附属仁济医院)、黄慧强(中山大学附属肿瘤医院)、黄琼(复旦大学附属华山医院)、金洁(浙江大学医学院附属第一医院)、李建勇(江苏省人民医院)、李小秋(复旦大学附属肿瘤医院)、马军(哈尔滨血液病肿瘤研究所)、沈建箴(福建医科大学附属协和医院)、沈志祥(上海交通大学医学院附属瑞金医院)、宋玉琴(北京大学肿瘤医院)、王琳(四川大学华西医院)、颜晓菁(中国医科大学附属第一医院)、袁成录[山东大学齐鲁医院(青岛)]、张春雷(北京大学第三医院)、张薇(北京协和医院)、赵东陆(哈尔滨血液病肿瘤研究所)、朱军(北京大学肿瘤医院)、邹德慧(中国医学科学院血液病医院)、邹立群(四川大学华西医院)
中国临床肿瘤学会(CSCO)淋巴瘤专家委员会.常见原发性皮肤T细胞淋巴瘤诊疗中国专家共识(2024年版)[J].白血病·淋巴瘤,2024,33(6):321-328. DOI:10.3760/cma.j.cn115356-20240520-00067.
所有作者声明无利益冲突
























