
发作性运动诱发性运动障碍是发作性运动障碍中最常见的类型,主要表现为突然运动或姿势改变诱发的舞蹈症和肌张力障碍等运动障碍,卡马西平和奥卡西平可以有效控制发作。该病是一种常染色体显性遗传病,致病基因有PRRT2和TMEM151A。若临床医生对该病认识不足,易将其误诊为癫痫或癔症,延误治疗。本文对该病的病因及发病机制、流行病学、临床表现、辅助检查、诊断及鉴别诊断、治疗与预后等方面进行介绍。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
经全国继续医学教育委员会批准,本刊开设继教专栏,2024年共刊发10篇继教文章,文后附5道单选题,读者阅读后可扫描标签二维码答题,每篇可免费获得Ⅱ类继教学分0.5分,全年最多可获5分。本年度继教答题得学分活动将于10月20日结束。
发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD;OMIM 128 200)最早于1967年被报道并命名为发作性运动诱发性舞蹈手足徐动症(paroxysmal kinesigenic choreoathetosis,PKC),表现为在静止状态下突然运动后出现短暂性不自主的舞蹈样动作[1]。此后,人们发现这些患者还会出现肌张力障碍等症状,因此将其重新命名为PKD[2],并对发作性运动障碍进行临床分类,发现PKD是最常见的类型。本病可以治疗,卡马西平和奥卡西平能有效控制发作,但由于PKD是罕见病,患病率低,临床医生易将其误诊为癫痫、癔症或其他发作性疾病,因此早期正确识别PKD具有重要的临床意义。
PKD分为原发性和继发性两种[3],临床上绝大多数患者是原发性PKD,多呈常染色体显性遗传,目前已鉴定出2个致病基因,包括PRRT2和TMEM151A[4, 5],少数KCNA1基因突变也可导致PKD表型[6, 7],存在外显不全现象[8]。继发性PKD相对少见,主要是由其他疾病如多发性硬化、肿瘤、外伤、钙化、血管病等导致基底节病变进而引发PKD样发作[9, 10, 11]。
PKD的首个致病基因PRRT2被鉴定报道后,国内外学者对PRRT2蛋白的生理功能及PRRT2基因突变的致病机制进行了大量研究。有研究结果表明,PRRT2基因编码的PRRT2蛋白是特异性表达于神经元的膜蛋白,可以通过与钠离子通道以及钠钾泵相互作用影响神经元的兴奋性[12],还可以通过与突触相关蛋白相互作用影响突触传递的过程[13]。当PRRT2基因突变时,体内PRRT2蛋白的缺乏会加速钠离子通道从失活状态恢复,增强神经元的兴奋性,从而促进小脑皮质去极化扩布(spreading depolarization,SD)的产生[14],SD事件和运动障碍的发生密切相关(图1)。此外,对PKD患者的颅脑影像学研究结果表明,除小脑外,基底节、丘脑、皮质等脑区也有明显异常,但这些脑区在PKD发作过程中起到的作用并不明确。因此,PKD的发病机制仍有待进一步阐明。
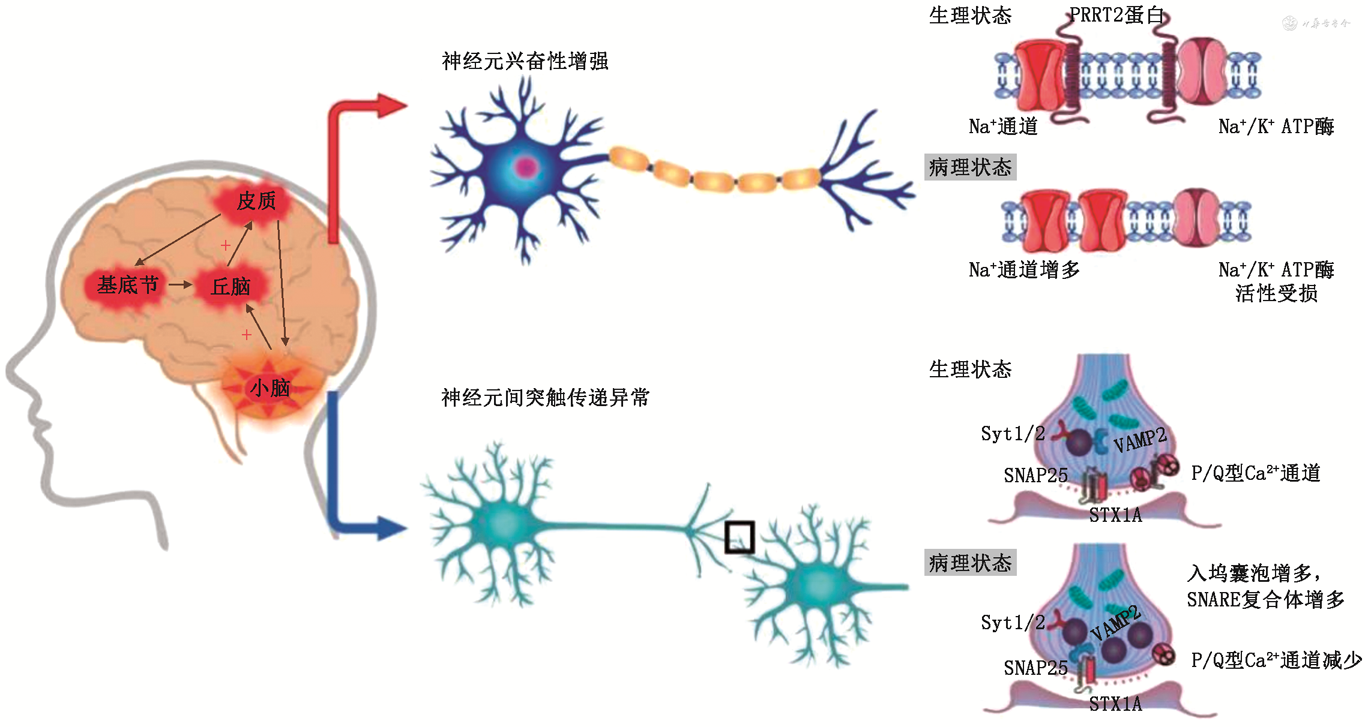
VAMP2:突触小泡膜蛋白;SNAP25:相对分子质量为25 000的突触关联蛋白;STX1A:突触融合蛋白1A;Syt1/2:突触结合蛋白1/2;SNARE:可溶性N-乙基马来酰亚胺敏感的融合蛋白
PKD是一种罕见病,目前尚无确切的患病率数据,粗略估计为1/150 000(https://www.ncbi.nlm.nih.gov/books/NBK1460/)。随着对PKD致病基因PRRT2和TMEM151A的报道[4, 5],临床医生对PKD的识别能力有所提升,这有助于未来更准确地评估其流行病学特征。
1.发病年龄:PKD通常在儿童期或青春期发病,7~15岁发病者最常见[15]。少数患者可早至出生后数月,或晚至20~30岁发病[16]。男性患者较女性患者多见,比例约为(2~4)∶1,但导致这种性别差异的原因迄今不清。
2.发作诱因:静止状态下突然运动是引起PKD发作最主要的诱因,比如从坐位突然起立、突然起跑、过红绿灯时突然起步、上下公交车等。此外,运动过程中突然加速、突然改变运动幅度亦可出现发作。部分患者改变身体姿势也会诱导发作[17]。情绪紧张、担心、受到惊吓或过度通气时通常更容易出现发作。
3.发作先兆:70%~80%的患者在发作前会出现先兆表现,预感到即将出现发作[3,16],常表现为一种难以描述的感觉,部分患者描述为肢体的麻木、无力或针刺感。患者可在先兆表现出现时,通过放慢动作或保持不动等,有意识地阻止发作。
4.发作形式:PKD发作主要表现为全身不自主的舞蹈样动作、肢体僵硬、偏身投掷症、手足徐动症等[18]。发作可为单侧或双侧。部分患者可累及面部,出现表情异常、讲话或发声困难等[15]。同一例患者发作的严重程度也会轻重不一,严重时可累及全身,轻微时仅累及单侧肢体。
5. 发作持续时间和频率:PKD患者在发作时意识完全清楚,发作持续时间短暂,超过95%的患者在1 min内可自行缓解[3,16]。患者的发作频率变化较大,与患者的活动情况有一定关系,多则每天数十次,少则每月数次甚至每年数次,大部分患者为每天1~20次,发作频率常在青春期达到高峰,此后可随年龄增大,发作频率逐渐减少,部分患者可在30岁以后自然缓解,不再发作。
6.伴随症状:部分患者可伴随其他症状,这可能与患者携带的基因突变有关。例如,部分携带PRRT2杂合突变的患者可伴随自限性婴儿癫痫或良性家族性婴儿癫痫、热性惊厥、偏瘫性偏头痛等[19],而携带PRRT2纯合突变或16p11.2染色体微缺失的患者,可出现语言和运动发育迟缓、智力障碍、自闭症、肥胖、脊柱畸形、头围增大和先天性心脏病等表现[20, 21]。
7.体格检查:发作间期PKD患者的神经系统体检通常无阳性体征。高抬腿试验常可诱发患者出现发作。
临床诊断PKD主要依赖典型的临床表现和诱发试验,患者的实验室和影像学检查一般无明显异常,以下检查主要用于排除其他可能引起类似发作的疾病。
1.血液生化:对于初次发病且无家族史的儿童,应完善相关血液生化和代谢筛查,以排除代谢性疾病导致的运动障碍。
2.脑电图:尽管PKD患者的脑电图通常正常,但在发作期间或触发运动后立即进行脑电图检查可能捕捉到异常放电模式,有助于区分其他发作性疾病,如癫痫。
3.颅脑影像学:颅脑MRI有助于排除继发性PKD,如多发性硬化、基底节钙化、颅内占位、脑血管畸形等引起的PKD样发作。
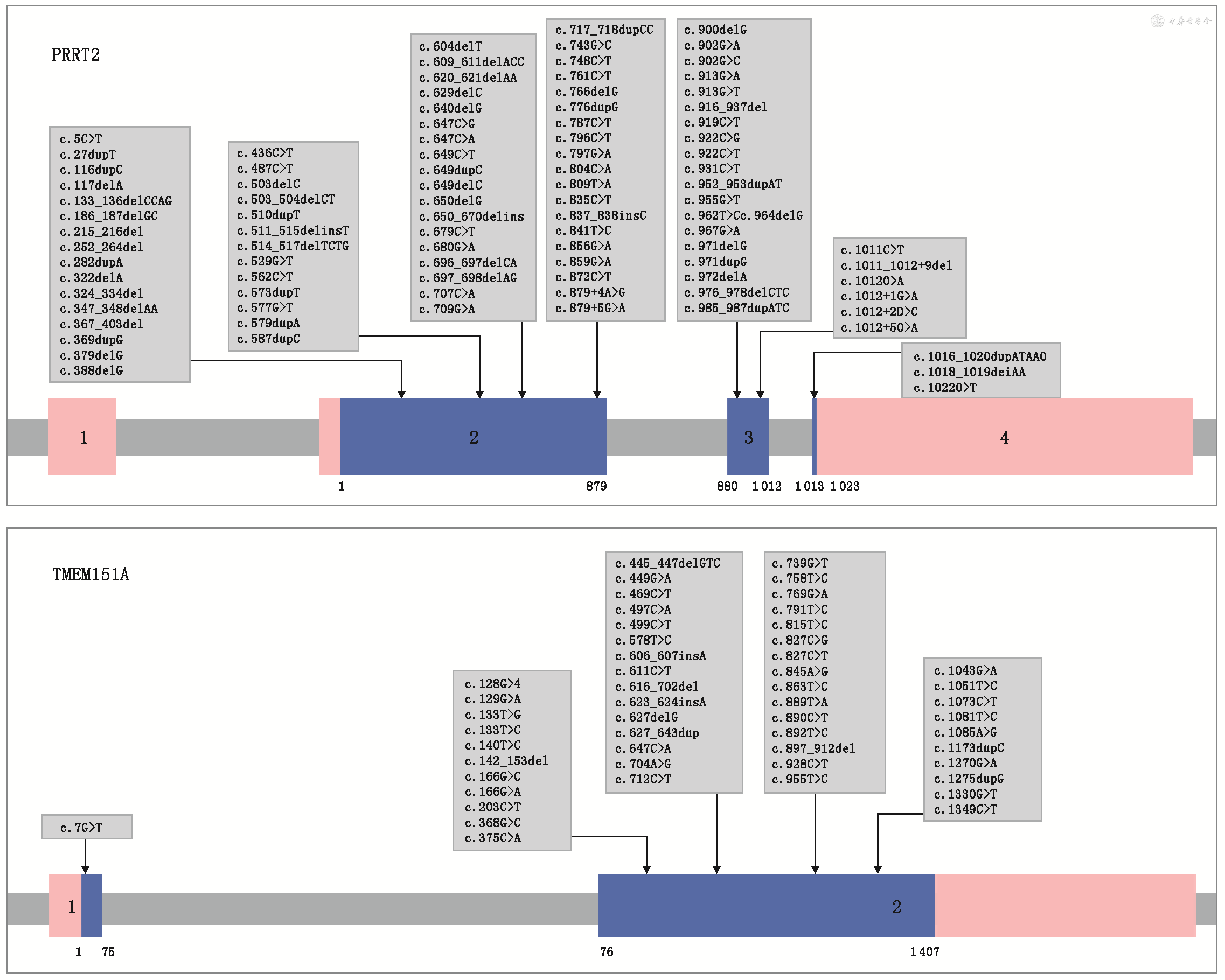
4.基因检测:对有家族史的PKD患者应进行基因检测,包括筛查PRRT2、TMEM151A和KCNA1等基因突变。目前已报道的PRRT2及TMEM151A致病突变详见图2。大部分PKD患者携带PRRT2基因的c.649dupC突变,建议优先筛查该突变。若上述基因检测未见异常,应进一步筛查PRRT2基因所在区域(16p11.2)的大片段缺失[21, 22]。
2022年我国发表了《中国发作性运动诱发性运动障碍诊治指南》[23],PKD诊断标准如下:(1)突然运动或改变体位诱发的不自主运动,包括舞蹈症、肌张力障碍、手足徐动症等,持续时间一般不超过1 min,发作时意识清晰,发作间期神经系统体检正常;(2)排除其他脑部病变等继发性因素;(3)小剂量卡马西平或奥卡西平疗效显著;(4)基因检测确定患者携带PRRT2或TMEM151A致病变异。临床诊断PKD:符合(1)~(3);基因诊断PKD:符合(1)~(4);携带者:符合(4)。
1.局灶性癫痫:无意识障碍的局灶性癫痫极易和PKD发作混淆。二者都有发作性、短暂性、重复性、刻板性且抗癫痫药物治疗有效的特点。但是PKD发作前有明显且单一的运动诱因,发作时意识清楚,多可正常对答,发作后对发作情境可完全回忆。脑电图是鉴别两种疾病的重要辅助手段,PKD患者发作间期脑电图正常,而癫痫患者的脑电图多有痫样放电。
2.其他类型的发作性运动障碍:包括发作性非运动诱发性运动障碍(paroxysmal nonkinesigenic dyskinesia,PNKD)和发作性持续运动诱发性运动障碍(paroxysmal exercise-induced dyskinesia,PED)。它们的发作症状与PKD相似,但是诱因、持续时间、致病基因和抗癫痫药物治疗反应均与PKD不同。PNKD由非运动因素诱发,包括咖啡、酒精、月经来潮、疲劳等,每次发作持续数分钟至数小时,致病基因为MR-1[24](现已更名为PNKD基因);PED多由长时间持续运动后诱发,每次发作持续数分钟至数十分钟,致病基因为SLC2A1(GLUT1)[25]。抗癫痫药物对PNKD和PED无明显疗效。
3.先天性肌强直:当PKD患者以肌张力障碍为主要表现时可能与先天性肌强直混淆,后者对抗癫痫药物如卡马西平治疗也有一定效果,遗传模式也是常染色体显性遗传[26],但肌电图常有肌强直电位,而PKD患者的肌电图正常。
4.发作性共济失调Ⅰ型(episodic ataxia type 1,EA1):KCNA1是EA1的致病基因,但少数KCNA1基因突变也可导致PKD表型[6, 7],而且EA1发作的常见诱因也是运动。然而,EA1发作时主要以共济失调表现为主[27],包括头晕、构音障碍、行走不稳等,情绪紧张、惊吓等亦可诱发,多持续数秒到数分钟,由此可与PKD相鉴别。
5.继发性PKD样发作:多发性硬化、肿瘤、外伤、钙化、血管病等引起的基底节病变,以及系统性红斑狼疮、甲状腺功能减退/亢进、糖代谢异常等系统性疾病亦可导致PKD样发作,临床上需根据病史和辅助检查等进行鉴别,避免漏诊和误诊。
6.心因性非癫痫发作(psychogenic non-epileptic seizure,PNES):PNES由心理因素所致,情绪紧张或某些环境因素可诱发,发作时脑电图正常。PKD患者在发作间期神经系统体检及相关辅助检查多为正常,因此易被误诊为PNES。但是PNES发作时间长且抗癫痫药物治疗无效,以此可与PKD鉴别。
对于临床诊断或基因诊断确诊的PKD患者,建议口服小剂量卡马西平或奥卡西平,卡马西平推荐剂量50~200 mg/d,每天分1~2次口服[28],奥卡西平推荐剂量为150~300 mg/d[29]。口服卡马西平或奥卡西平前,建议检测HLA-B1502等位基因,避免发生Steven-Johnson综合征等严重并发症。若患者对卡马西平或奥卡西平过敏,可选择拉莫三嗪、左乙拉西坦、托吡酯等抗癫痫药物[16],这些是治疗PKD的二线药物。部分PKD患者可能存在自卑、焦虑、抑郁等情绪反应[30],应给予适当的心理疏导和相关药物治疗。由于大部分PKD患者在30岁后症状有自发缓解趋势,预后良好,无需终身治疗。对于继发性PKD患者,应针对病因进行治疗。
PKD是发作性运动障碍中最常见的一种类型,容易误诊为癫痫、癔症或其他发作性疾病,临床上应注意鉴别。由于该病对卡马西平等抗癫痫药物治疗效果良好,早期识别十分关键。临床上绝大多数患者是原发性PKD,建议完善PRRT2、TMEM151A和KCNA1等基因检测,若患者携带基因突变,对于指导用药及遗传咨询具有重要意义。
吴志英, 李宏福. 发作性运动诱发性运动障碍[J]. 中华神经科杂志, 2024, 57(9): 1020-1024. DOI: 10.3760/cma.j.cn113694-20240324-00187.
所有作者声明无利益冲突
None declared
1.发作性运动诱发性运动障碍最常见的致病基因是?
A.PMP22
B.PRRT2
C.Parkin
D.PRNP
2.发作性运动诱发性运动障碍首选的治疗药物是?
A.丙戊酸钠
B.苯巴比妥
C.卡马西平
D.左乙拉西坦
3.发作性运动诱发性运动障碍首选的高发年龄是?
A.7~15岁
B.20~30岁
C.30~50岁
D.60岁后
4.以下哪项不是发作性运动诱发性运动障碍的常见临床表现?
A.舞蹈症
B.肌张力障碍
C.手足徐动
D.肌阵挛
5.以下哪项符合发作性运动诱发性运动障碍的特征?
A.发作时意识丧失
B.头颅MRI常有异常
C.脑电图常有痫样放电
D.发作时间常小于1 min

























