患者男,27岁,因“无明显诱因出现剧烈胸痛伴心悸、气短、胸闷”于2018年8月收治入西京医院。计算机断层扫描血管造影检查结果提示主动脉A型夹层。基于二代测序的遗传性主动脉疾病相关的15个基因的组合检测及Sanger测序验证发现患者COL3A1基因存在c.998G>T(p.Gly333Val)杂合错义突变。通过家系成员的Sanger测序验证,突变c.998G>T与该家系患者表型共分离。根据ACMG遗传变异分类标准与指南判读,该突变致病等级为“可能致病”,携带该突变可明确诊断为“血管型Ehlers-Danlos综合征”。明确诊断后,对患者进行了对症治疗,但病情发展迅速,患者放弃治疗,出院后不久死亡。本研究报道了1个COL3A1基因的新突变,扩展了该基因的突变谱。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
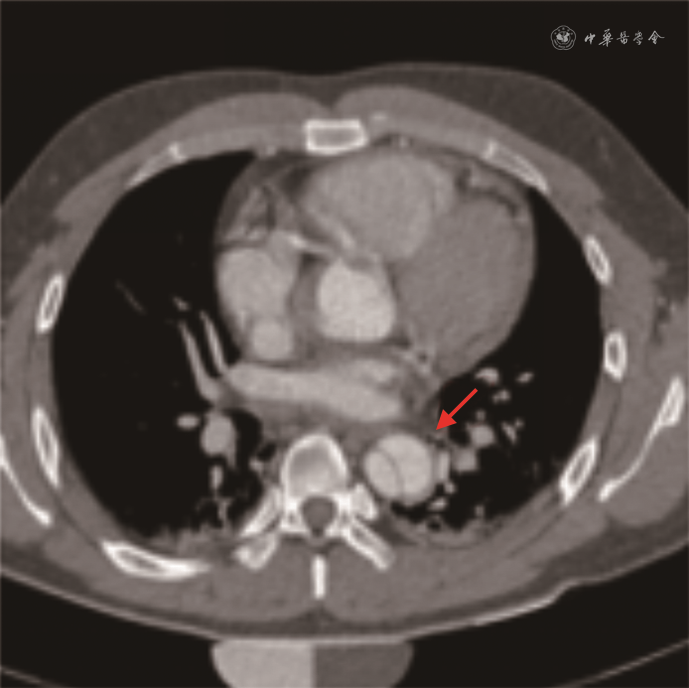
患者男,27岁,体重75 kg,身高170 cm,2018年8月因胸痛就诊于空军军医大学附属西京医院心血管外科。患者于15 h前无明显诱因感胸背部剧烈疼痛,伴心悸、气短、胸闷,无发热,无咳嗽、咳痰,无痰中带血,无腹痛、腹胀,无恶心、呕吐,无呕血、便血,能平卧,无夜间阵发性呼吸困难,无下肢水肿,无头晕、晕厥。面容正常,表情自如,神志清楚,查体合作。查体:体温36.5 ℃,脉搏78次/min,呼吸14次/min。心前区饱满,心尖搏动未见异常,位于胸骨左锁骨中线内侧1.5 cm。触诊心尖搏动位置同前,左锁骨中线内侧1.5 cm,未触及震颤,未触及心包摩擦感。心脏相对浊音界不大。听诊心率78次/min,律齐,未闻及早搏,心音可,各瓣膜区听诊未闻及病理性杂音。无异常血管征。血压126/71 mmHg(1 mmHg=0.133 kPa),否认高血压病史。吸烟15年,平均10支/d。计算机断层扫描血管造影术检查提示主动脉A型夹层(图1)。
基因组合测序:经患者及其家属知情同意后,取患者、患者父亲及患者弟弟静脉血各4 ml,乙二胺四乙酸(ethylene diamine tetraacetic acid,EDTA)抗凝。采用西安天隆科技有限公司全血基因组核酸提取试剂盒常规提取外周血样本DNA,采用Qubit 2.0型荧光计及1.0%的琼脂糖凝胶电泳进行质检,全部DNA样本质量符合测序要求,于‒20 ℃冰箱冻存。通过OMIM、HGMD、Clinvar 等数据库的查询和比较,同时参考 Gene review 及主动脉疾病相关指南,选取15 个与遗传性主动脉疾病相关的基因,对先证者基因组DNA进行该15个基因的组合检测,包括FBN1、FBN2、TGFB2、TGFBR1、TGFBR2、SMAD3、SMAD4、MYH11、ACTA2、MYLK、COL3A1、SLC2A10、NOTCH1、SKI及PRKG1基因的外显子区及外显子-内含子交界处。文库构建采用Thermo Scientific公司Ion AmpliSeqTM Library Kit 2.0试剂盒,使用Ion Torrent PGM DX(Thermo Scientific公司)平台进行序列分析。使用序列比对软件将测序序列与参考基因组GRCh37/hg19比对;然后使用VEP软件分析出单核苷酸多态性(single nucleotide polymorphism,SNP)、插入缺失;最后根据测序深度、突变质量对检测到的SNP、插入缺失进行过滤和筛选,得到高质量可靠的突变。滤除不影响基因功能的同义突变和公共遗传突变数据库(dbSNP数据库、千人基因组数据库、ExAC 数据库和 gnomAD数据库)中正常人携带的常见变异(最小等位基因频率>0.1%),对文献及OMIM、HGMD、ClinVar等多种公共数据库中记录的与遗传性主动脉疾病相关的候选基因上的突变进行筛选。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南中对突变位点致病性的分类标准,从致病性的5个类别:致病、可能致病、意义不明确、可能良性和良性中确定1个分类,因可能良性及以下级别突变的致病风险<5%,所以本研究筛除良性突变和可能良性突变,筛选出致病性分级为致病、可能致病和意义不明的突变[1]。该研究获得西京医院伦理委员会批准(KY20202095-F-1)。
Sanger测序验证患者及家属的变异位点:取患者及其父亲、弟弟外周血基因组DNA,针对COL3A1(NM_000090)可疑致病位点,采用引物设计软件Oligo 7设计PCR扩增引物(正向5′-GCCTGCACTAAGTCACAGAATTCA-3′,反向5′-AATCAGTTGCAAGAAGCAATCTAATGC-3′),经加利福尼亚大学圣克鲁兹分校基因组学研究所数据库(University of California Santa Cruz,UCSC)In silico PCR验证引物序列的可靠性,由西安擎科泽西生物科技有限责任公司合成。采用PCR扩增候选位点所在的DNA序列,取PCR产物2 μl经琼脂糖凝胶电泳检测,扩增成功,对先证者及家系成员进行Sanger正反向测序,测序结果与UCSC数据库人类基因正常序列对比。
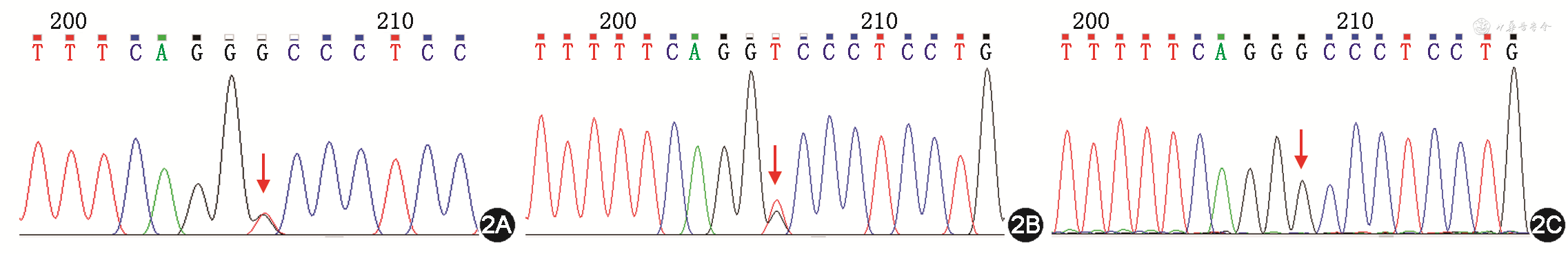
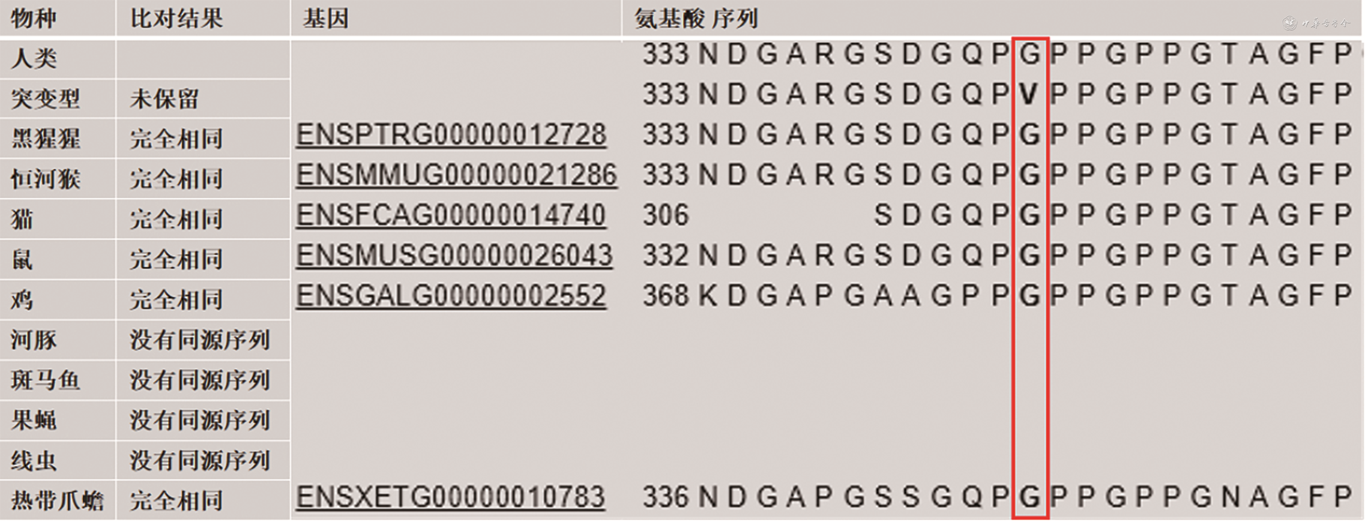
基因组合检测及Sanger测序发现患者的COL3A1基因存在c.998G>T(p. Gly333Val)杂合突变(图2A),即COL3A1基因编码区第998位碱基由G突变为T,导致编码的Ⅲ型胶原蛋白序列发生改变,即第333位氨基酸由甘氨酸(Gly)突变为缬氨酸(Val)。Sanger测序显示此家系中患有主动脉夹层动脉瘤的弟弟也携带此变异(图2B),而体健的父亲未携带此变异(图2C),按照孟德尔遗传规律,推测变异遗传自腹膜后血管破裂去世的母亲(家系图谱见图3),这一基因型-表型关联性符合家系共分离(pathogenic supporting 1,PP1[2]);生物学软件如尺度不变特征变换(0)、多态性表型v2(1.0)、Mutation Taster(1.000)等多个软件预测该突变位点是“有害的”(pathogenic supporting 3,PP3),Mutation Taster中的保守性分析提示该位点在多个物种间是保守的(图4);同一氨基酸位点至少存在1个不同氨基酸改变的可能致病变异p.Gly333Asp(pathogenic moderate 5,PM5);根据ExAC数据库、千人数据库、gnomAD 数据库分析,该变异位点的人群频率暂无记录(pathogenic moderate 2,PM2)。根据美国医学遗传学与基因组学会遗传变异分类标准与指南,c.998G>T突变评级为可能致病(证据为PP1+PP3+PM5+PM2)。结合患者临床表现,确诊为血管型Ehers-danlos综合征(Ehers-Danlos syndrome,vascular type,vEDS)。患者入院后病情进展较快,家属放弃治疗自动出院,出院后随访,患者死亡。本研究中携带突变的先证者弟弟,在进行家系遗传咨询时已告知定期随访,密切观察病情变化,并因其已婚未孕,告知其备孕前进行产前遗传咨询。患者1岁10个月的女儿因年龄尚小未进行基因检测,已告知成年后随访检测。
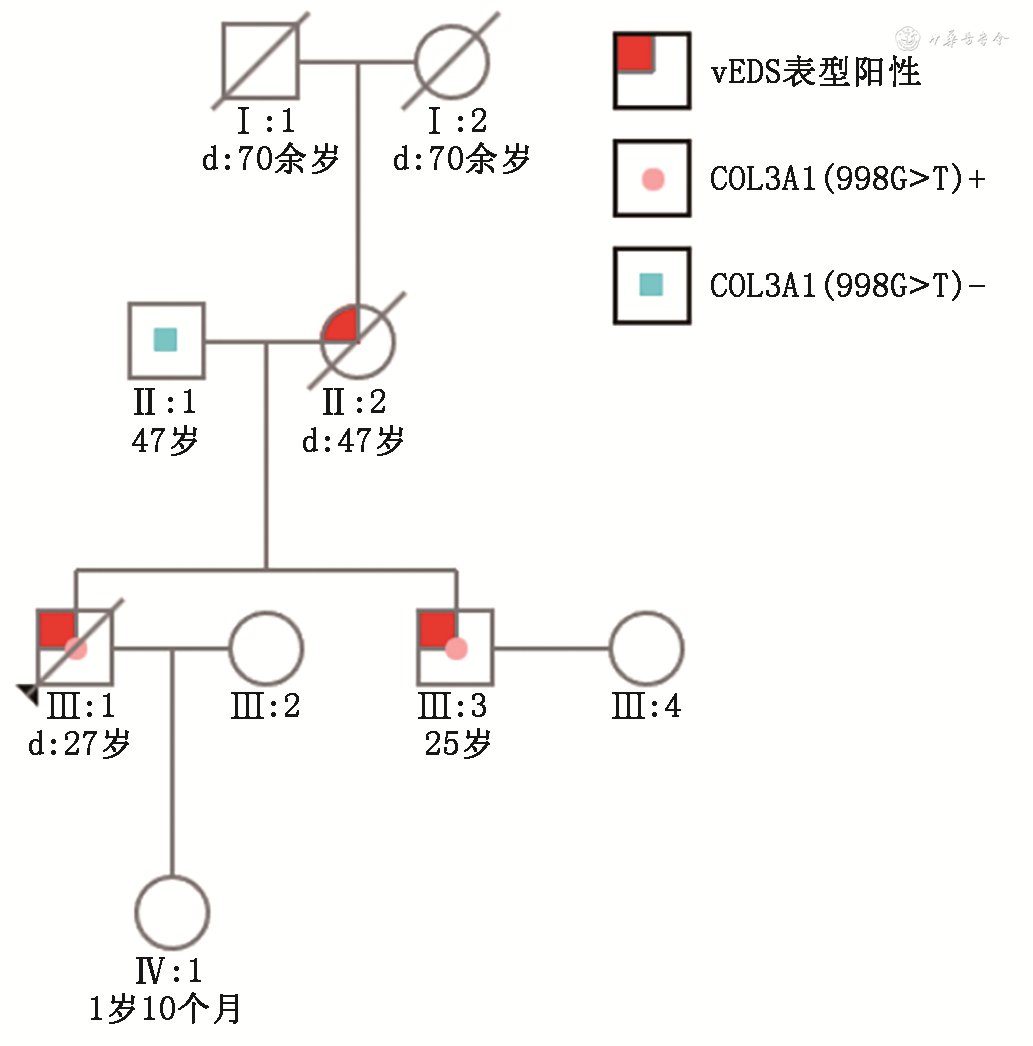
注:vEDS为血管型Ehers-danlos综合征。圆圈代表女性,正方形代表男性,箭头为先证者,斜线表示已死亡
EDS是一类具有不同遗传病因的遗传性结缔组织疾病[3],以皮肤弹性过度、关节活动度大及组织脆弱为主要临床特征。EDS具有13种临床不同的亚型[4],vEDS为EDS Ⅳ型,是并发症最为严重的一种,发病率为1/250 000~1/100 000[5],占EDS总数不足4%[6]。COL3A1基因发生突变是vEDS的主要致病原因[7],其遗传模式为常染色体显性遗传,约50%患者由新发突变引起。基因检测出COL3A1的致病性变异,则可确诊为vEDS[8, 9]。已确诊为vEDS的患者,首选CTA或MRA等无创的影像学方法以明确血管病变及随访观察,介入性的血管造影被视为禁忌[10]。vEDS病死率很高,自发性动脉、肠道和/或子宫破裂是其主要死亡原因。
先证者与其患有主动脉夹层动脉瘤的弟弟均携带此变异,而他们体健的父亲未携带此变异。根据孟德尔遗传规律推测,该变异遗传自母亲,先证者母亲在先证者就诊前一个月在当地医院因腹膜后血管破裂去世。先证者外公、外婆均于70多岁自然死亡,推测该突变系先证者母亲的新发突变。COL3A1基因编码Ⅲ型前胶原蛋白α-1链,其组装成Ⅲ型胶原蛋白的三聚体分子。作为血管和空腔器官的主要结构成分,Ⅲ型胶原减少或异常会导致自发性动脉或器官破裂的风险增加。25%的vEDS患者在20岁前发生动脉、肠道或其他空腔器官的并发症,超过80%的患者会在40岁前至少发生一次并发症;70%的血管型EDS患者表现为血管破裂或其他器官破裂,而发生第一次大动脉或胃肠道并发症的平均年龄仅为23岁[11]。患有vEDS的患者中位存活年龄48岁[12]。由于皮肤和韧带组织内Ⅲ型胶原蛋白仅占15%,vEDS患者皮肤和关节病变并不典型,本研究中的几位vEDS患者均无皮肤和肢体特征,如其他EDS亚型常见的薄而半透明的皮肤、广泛的瘀伤及小关节活动过度等表现。研究表明[13],COL3A1变异的类型影响vEDS的病程及患者的临床表型,杂合错义突变比剪接突变及单倍剂量不足突变的致病性严重程度更高,患者初次诊断的年龄更低,这可能因为COL3A1基因编码的Ⅲ型胶原蛋白是三聚体分子结构,杂合错义突变导致任一单体的错误表达,都会导致整个蛋白功能降低或缺失,即显性负效应机制[14]。其中错义突变导致的甘氨酸残基被取代引起的疾病的严重程度最高,尤其是在被缬氨酸残基取代的情况下,这可能与取代后的氨基酸残基极度不稳定有关[13]。本研究报道的vEDS家系中,先证者25岁即发生急性、恶性的并发症,也证实了这一理论。
vEDS目前尚无根治办法[15, 16],治疗和管理的重点是预防疾病进展和随后的并发症,需要采用多学科方法,各学科专家通常针对具体临床表现对症治疗。已确诊为vEDS的患者,首选计算机断层扫描血管造影术或核磁共振血管造影术等无创的影像学方法以明确血管病变及随访观察,不推荐介入性的血管造影,因vEDS患者的血管脆性极高,术中导管进入处可发生血管壁撕裂及夹层,注射压力亦可导致动脉瘤形成[7]。对明确诊断患有vEDS的患者,需定期进行心血管筛查,以尽可能减少风险因素。高血压会增加本已脆弱的血管系统的压力,并随着时间的推移增加并发症的风险,因此应通过药物治疗积极控制血压(收缩压<120 mmHg)和心率(<70 次/min),控制血管疾病高危因素(如高脂血症、吸烟、饮酒)[17]。对伴有动脉瘤患者,建议每3~6个月复查超声心动图、动脉核磁共振血管造影术或计算机断层扫描,对不伴有动脉瘤患者则可延长至每12个月随访1次。本研究中,先证者弟弟已确诊伴有主动脉夹层动脉瘤,在进行遗传咨询时告知本人定期随访,密切观察病情变化,并因其已婚未孕,告知其备孕前进行产前遗传咨询。患者1岁10个月的女儿因年龄尚小未进行基因检测,已告知成年后随访检测。
vEDS罕见,易误诊及漏诊,需与其他具有重叠临床表型的遗传性主动脉疾病进行鉴别诊断,包括马凡综合征等相关疾病。临床医生仅通过患者临床表现往往难以确诊,特别是表型不确定的患者,基因检测可明确诊断、指导疾病的治疗,具有重要意义。本研究报道了1例vEDS患者及家系的基因检测,丰富了COL3A1基因-表型相关性图谱,明确了该家系中出现多例血管受累患者的遗传学病因,为vEDS疾病基因-疾病相关性提供了依据,加深了临床医生对罕见病的认识。
李金洁, 杨柳, 辛毅娟, 等. COL3A1基因新突变导致血管型Ehlers-Danlos综合征一家系的临床特征与遗传学分析[J]. 中华检验医学杂志, 2024, 47(9): 1082-1085. DOI: 10.3760/cma.j.cn114452-20231214-00358.
所有作者声明无利益冲突


























