
线粒体病是一组遗传代谢病,因线粒体结构和(或)功能异常造成能量代谢功能损害,可导致单器官或多器官功能障碍。在新生儿期发病的线粒体病为新生儿线粒体病。新生儿线粒体病致病基因复杂,患儿表型差异显著,常常病情凶险,缺乏特异和敏感的生物标志物,且治疗方法有限,诊断及救治极具挑战性。为进一步规范新生儿线粒体病的诊断与治疗,新生儿、神经、代谢内分泌及遗传学等专家基于国内外临床证据,结合实践经验,从概念、定义、早期识别、生化代谢、影像、基因检测、病理、治疗、遗传咨询及产前诊断等层面对新生儿线粒体病进行论述,并形成诊疗专家共识。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
线粒体病是罕见病中相对常见的疾病,是由于编码线粒体结构蛋白或参与线粒体功能蛋白的基因变异引起的一组遗传病,通常存在一个或多个电子传递链复合物功能障碍或其他线粒体功能和(或)结构紊乱,从而导致线粒体能量代谢障碍。线粒体病可因核DNA(nuclear DNA,nDNA)或线粒体DNA(mitochondrial DNA,mtDNA)变异引起,可在任何年龄发病,总发病率估计为1/4 300[1, 2],儿童发病率约为1/6 000,其中5%~30%的患儿在新生儿期出现症状,且绝大部分在生后48 h内发病[3]。
新生儿线粒体病发病率低,临床表现缺乏特异性,易被误诊、漏诊。尽管国内外陆续发表了许多有关新生儿线粒体病的个案报道,但目前尚无关于疾病诊治的共识或指南。另外,我国各地诊疗水平存在差异,造成不同医疗机构对新生儿线粒体病的管理缺乏同质化,疾病转归差异较大。为此,执笔组邀请新生儿、神经、代谢内分泌及遗传学专家讨论,以国内外循证医学证据为基础,结合临床实践经验制订了本共识。本共识检索的数据库包括PubMed、Embase、Web of Science、Cochrane Library、Medline、中国知网、万方数据、维普和中华医学期刊全文数据库,系统检索了从建库至2023年12月的中英文文献。本共识的目标人群为新生儿科医护人员及相关领域的研究人员,以期进一步规范新生儿线粒体病的诊断与治疗。
线粒体是细胞内的一种双膜细胞器,是能量与物质代谢中心,除成熟红细胞外的所有细胞中均有线粒体存在。线粒体通过5个电子传递链复合物生成三磷酸腺苷,提供细胞所需的大部分能量,参与细胞凋亡、活性氧产生、钙稳态和免疫调节等活动。线粒体受nDNA与mtDNA的双重调控,成年发病的线粒体病患者中约80%为mtDNA致病变异所致,儿童期发病的线粒体病患儿约80%为nDNA致病变异[4]。
线粒体病既可原发,也可继发。原发性线粒体病是由于nDNA或mtDNA的致病性变异直接影响呼吸链成分和呼吸链功能,或线粒体自身功能和(或)结构异常,导致单器官或多器官功能障碍。继发性线粒体病是指由于其他疾病导致线粒体损伤而引起的一系列疾病[1]。本共识主要针对新生儿期发病的原发性线粒体病。
推荐意见1 新生儿线粒体病表型缺乏特异性,少数患儿胎儿期生长受限、胎动减少,出生后可表现为喂养困难、肌张力异常、癫痫发作、肝功能损害、心肌损害、呼吸循环衰竭。新生儿出现以上多系统表现时应警惕线粒体病的可能。
推荐说明 线粒体病可表现为任何症状、任何器官或组织损害、任何年龄发病、任何遗传模式,尤其易影响高能量需求组织,如心脏、骨骼肌和中枢神经系统[5]。新生儿线粒体病患儿表型缺乏特异性,约1/3的患儿在宫内可能出现胎儿生长受限、羊水过多或减少、胎动减少等表现,偶尔有胎儿肥厚型心肌病、心律失常及胎儿水肿,出生后可表现为喂养困难、肌张力低下、癫痫发作、肝功能损害(伴或不伴胆汁淤积、低血糖)、呼吸功能不全及循环衰竭[3,6, 7],常被考虑为围产期窒息、新生儿败血症、心力衰竭、坏死性小肠结肠炎等。新生儿线粒体病最常见的临床表现是肌张力低下、血乳酸升高,其次为呼吸暂停、高氨血症、心肌病和肝损伤[8]。心脏受累常表现为肥厚型心肌病、扩张型心肌病、心律失常及心力衰竭,这也是决定新生儿线粒体病预后的重要因素[8, 9, 10]。研究发现,生后2 d内出现线粒体病多系统功能受累预示病死率更高[7]。新生儿线粒体病除了可表现为单一器官或多系统受累,也可以表现为某种综合征,见表1。
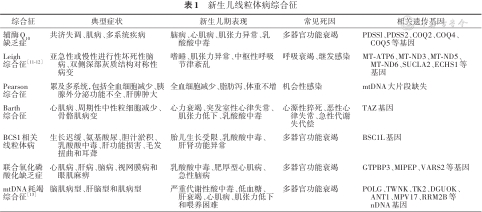
新生儿线粒体病综合征
新生儿线粒体病综合征
| 综合征 | 典型症状 | 新生儿期表现 | 常见死因 | 相关遗传基因 |
|---|---|---|---|---|
| 辅酶Q10缺乏症 | 共济失调、肌病、多系统疾病 | 脑病、心肌病、肌张力异常、乳酸酸中毒 | 多器官功能衰竭 | PDSSl、PDSS2、COQ2、COQ4、COQ5等基因 |
| Leigh综合征[11, 12] | 亚急性或慢性进行性坏死性脑病、双侧深部灰质结构对称性病变 | 嗜睡、肌张力异常、中枢性呼吸节律紊乱 | 呼吸衰竭、继发感染 | MT-ATP6、MT-ND3、MT-ND5、MT-ND6、SUCLA2、ECHS1等基因 |
| Pearson综合征 | 累及多系统,包括全血细胞减少、胰腺外分泌功能不全、肝脾肿大 | 全血细胞减少、脂肪泻、体重不增 | 机会性感染 | mtDNA大片段缺失 |
| Barth综合征 | 心肌病、周期性中性粒细胞减少、骨骼肌病变 | 心力衰竭、突发室性心律失常、肌张力低下、乳酸酸中毒 | 心源性猝死、恶性心律失常、急性代谢失代偿 | TAZ基因 |
| BCS1相关线粒体病 | 生长迟缓、氨基酸尿、胆汁淤积、乳酸酸中毒、肝功能损害、毛发扭曲和耳聋 | 胎儿生长受限、乳酸酸中毒、肝肾功能异常 | 多器官功能衰竭 | BSC1L基因 |
| 联合氧化磷酸化缺乏症 | 心肌病、肝病、脑病、视网膜病和眼肌麻痹 | 乳酸酸中毒、肥厚型心肌病、急性脑病 | 多器官功能衰竭 | GTPBP3、MIPEP、VARS2等基因 |
| mtDNA耗竭综合征[13] | 脑肌病型、肝脑型和肌病型 | 严重代谢性酸中毒、低血糖、肝衰竭、心肌病、肌张力低下和喂养困难 | 多器官功能衰竭 | POLG、TWNK、TK2、DGUOK、ANT1、MPV17、RRM2B等nDNA基因 |
识别新生儿线粒体病的临床表型仅为诊断评估的第一步,还需要各种检验及检查来辅助诊断。
推荐意见2 疑似新生儿线粒体病的患儿,除常规进行实验室检验,还需检测血氨、乳酸、丙酮酸、酰基肉碱、氨基酸及尿有机酸等。但这些代谢标志物检测结果正常不能排除线粒体病。
推荐说明
1.常规检验项目:对所有怀疑新生儿线粒体病的患儿应全面评估电解质及酸碱平衡,其中代谢性酸中毒可能与乳酸盐积聚所致阴离子间隙增加有关,也可能与肾小管功能障碍有关。肝功能损害、低血糖、血肌酸激酶升高等可反映肝脏和肌肉组织受累。
2.血与脑脊液代谢标志物:主要包括血乳酸、丙酮酸、血氨、氨基酸、游离肉碱和酰基肉碱水平等[14]。血乳酸>3 mmol/L、乳酸/丙酮酸比值和丙氨酸升高可作为疑似线粒体病患儿相对敏感的初始检测指标[3,8,15]。但新生儿血乳酸水平升高也可由于呼吸衰竭、循环衰竭、坏死性小肠结肠炎、败血症和其他遗传代谢病导致,血乳酸水平正常也不能排除线粒体病的可能[16, 17]。如果血或脑脊液乳酸升高,同步检测相应标本乳酸/丙酮酸比值>25,对诊断线粒体病意义较大[14,18]。血甘氨酸、脯氨酸和苏氨酸升高以及瓜氨酸降低均提示新生儿线粒体病可能;线粒体功能障碍可造成尿素循环障碍而导致继发性高氨血症[3,17, 18]。对于血游离肉碱及酰基肉碱异常,目前尚无数据证实其对新生儿线粒体病的诊断价值[19]。
3.尿代谢标志物:新生儿线粒体病患儿尿乳酸通常增高。约31%的患儿尿有机酸检查出现三羧酸循环中间产物(如琥珀酸盐、富马酸盐、α-酮戊二酸盐)升高,这可能为线粒体功能障碍的非特异性标志物[15,17]。尿甲基丙二酸浓度升高可能是SUCLA1基因或SUCLG1基因相关疾病的诊断线索[8],需要排除钴胺素代谢缺陷和维生素B12缺乏症等疾病导致的甲基丙二酸尿症。尿3-甲基戊烯二酸升高提示Barth综合征及3-甲基戊烯二酸尿症[10,20]。
4.其他代谢标志物:生长分化因子15和成纤维细胞生长因子21是提示线粒体功能障碍的两种新的代谢标志物[21]。国外研究发现上述两种因子对线粒体翻译缺陷或mtDNA缺失引起的肌病型线粒体病的一级诊断最为敏感[22, 23, 24, 25],但国内目前仅部分单位将其用于科研,临床应用价值需进一步研究。
推荐意见3 疑似新生儿线粒体病患儿需检查心电图、超声心动图、头颅磁共振成像(magnetic resonance imaging,MRI)、视觉及听觉诱发电位等。根据临床表型酌情完善脑电图、肌电图、肌肉超声。
推荐说明 心肌损害是新生儿线粒体病常见的危重症之一,对所有怀疑新生儿线粒体病的患儿均需做心电图和超声心动图检查明确是否有传导阻滞和肥厚型心肌病[10]。新生儿线粒体病患儿的视神经、视网膜和内耳常受累,对疑诊患儿应进行眼科检查和听力评估[26, 27]。所有疑诊新生儿线粒体病且伴有神经症状的患儿均应检查颅脑MRI明确是否有胼胝体、脑白质、双侧半球病变,长T1、长T2信号,DWI高信号[14,28],如有条件,可进一步分析磁共振波谱,评估脑组织局部代谢及乳酸酸中毒的程度以协助诊断[14]。若患儿出现惊厥发作或脑病,需做脑电图观察有无棘波、尖波和棘-慢复合波。若怀疑肌病和(或)神经病变,建议进行肌电图明确肌源性或神经源性受损,并进行神经传导检测。肌肉超声对线粒体病的敏感度低,但特异度高,需要经验丰富的超声专科医生实施,可作为疑似新生儿线粒体病患儿的检查之一[3]。
推荐意见4 疑似线粒体病新生儿临床表型高度提示某种基因变异时,可先进行常见mtDNA变异和(或)nDNA变异靶向检测。对于急危重症需要全面检测线粒体相关疾病的新生儿,首选快速全外显子组测序(whole exome sequencing,WES)+mtDNA检测,必要时补充全基因组测序(whole genome sequencing,WGS)检测。
推荐说明 对于疑似新生儿线粒体病的患儿,分子遗传学检测非常重要[29],明确诊断可为部分患儿提供靶向治疗,减轻病情[30]。目前常用的基因检测方法包括Sanger测序和二代测序(next generation sequencing,NGS),其中NGS包括针对性Panel检测、WES和WGS。WES主要针对nDNA变异,可识别外显子区域蛋白质编码基因内的遗传变异,包括单核苷酸替代变异、插入、缺失和拷贝数变异,但对于嵌合变异、结构改变,例如染色体重排、重复扩增及深度内含子和调节区变异等则需要通过WGS检测。对于疑似线粒体病的患儿,WES在各类研究中的诊断率为35%~70%,目前尚无关于WGS对于线粒体病诊断率的系统评估[30]。对于依据临床表型高度怀疑由特定基因变异所致的疑似线粒体病患儿,可先进行常见mtDNA变异和(或)nDNA变异的靶向测序,但可能会延长确诊时间。因为疑似线粒体病的新生儿常为急症和危重症患儿,临床表型不典型,且儿童线粒体病约80%的病因为nDNA致病变异,故优先推荐快速WES检测,如经济条件允许,可同时检测mtDNA,或直接选择WGS。如临床高度怀疑新生儿线粒体病,外周血基因检测阴性,需选择尿液或肌肉组织、口腔拭子进行基因检测。
推荐意见5 基因检测结果阴性或疑似新生儿线粒病的患儿,可对肌肉或肝脏组织进行病理检查。对于诊断不明、疑似新生儿线粒体病的濒死患儿,建议留取血、尿样本,充分告知家属患儿死亡后代谢尸检的重要性,并在患儿死亡后争取代谢尸检。
推荐说明 对于成年人及青少年,肌肉组织的病理学检查是确诊线粒体病以及鉴别其他肌病的重要方法,其可观察线粒体的超微结构、量化氧化磷酸化功能、检测呼吸链复合物功能等[14],但是,病理学检查的局限性在于难以区分原发性线粒体病与继发性线粒体病,而且新生儿肌肉组织少,病情不稳定时更难获取标本,缺乏新生儿酶活性的正常参考范围,因此目前临床极少针对新生儿进行病理学检查。如果基因检测阴性或结果不确定,例如外周血基因检测出临床意义未明变异或低水平异质性,但临床高度怀疑线粒体病或神经肌肉病时,在医疗技术发达的地区可尝试进行病理学检查。除了肌肉组织,建议对受影响最严重的器官进行病理学检查,如对主要表现为肝病的患儿进行肝活检。如无法获得组织样本,也可尝试通过皮肤活检或口腔拭子进行电子传递链分析[3]。对诊断不明的危重或濒死新生儿应关注病因诊断。患儿死亡前或家属放弃前留取相关标本或死亡后进行代谢尸检非常重要,建议保存适量尿液、干血片、抗凝血及冷冻组织,例如未经甲醛溶液固定的皮肤、肝、脑、心肌、骨骼肌、肾等,进行生化、代谢、血氨基酸、酰基肉碱谱、尿有机酸谱和基因检测[30]。
推荐意见6 因线粒体病标准评分量表中的症状和体征在新生儿期表现不完全,组织形态学检查不适合新生儿,故不推荐常规使用线粒体病标准评分量表进行线粒体功能障碍评估。
推荐说明 目前对于成年人和青少年推荐使用线粒体病标准评分量表,从临床表型、生物学指标、肌肉组织活检、酶学等角度判断是否存在线粒体功能障碍,多参考Morava量表[31]、儿童神经线粒体病标准评分量表[15,32]、改良的Walker量表[33]、Nijmegen量表[34]等。但新生儿线粒体病临床表型缺乏特异性,且无法在新生儿患儿群体中普遍开展量表中的组织形态学检查,故不推荐常规使用线粒体病标准评分量表评估新生儿线粒体功能障碍。
推荐意见7 如果新生儿表现为多器官系统功能障碍,同时合并代谢标志物及头颅MRI异常,建议按照本共识流程进一步检测基因。见图1。
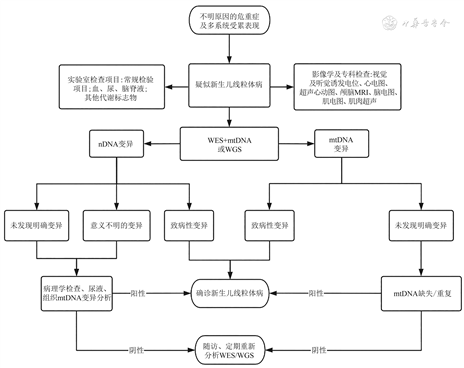
注:MRI为磁共振成像,nDNA为核DNA,WES为全外显子组测序,mtDNA为线粒体DNA,WGS为全基因组测序
推荐说明 新生儿线粒体病患儿的临床表现及生化、代谢等检查非特异,无单一的指标能证实患儿存在线粒体功能障碍,极易与其他新生儿危重症混淆。国内有许多新生儿线粒体病的个案报道,检索国内2013年4月至2024年4月的文献和专著,新生儿线粒体病患儿的主要临床表现为代谢性酸中毒、高乳酸血症、呼吸困难、吮奶差、反复低血糖、惊厥、黄疸、肝功能异常、呼吸暂停等,部分病例表现为脑白质病变、嗜睡、反应低下、肝病、呕吐、腹泻伴体重不增、营养不良、贫血[35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47]。国外Ebihara等[7]报道281例新生儿线粒体病患儿,主要表现为呼吸障碍(25.9%)、新生儿窒息(14.3%)、喂养困难/发育落后(10.6%)、先天性畸形(10%)、高乳酸血症(86%)。本共识专家组建议,对于新生儿出现不明原因的神经系统表现,例如惊厥、肌张力障碍、脑病、窒息等,同时合并1个或多个其他系统表现,包括内分泌系统(持续或反复低血糖、代谢性酸中毒、甲状腺功能低下)、消化系统(喂养困难、肝功能受损)、心血管系统(心肌病、心律失常)等,同时代谢标志物异常(持续高乳酸血症、高丙氨酸血症、高氨血症)或脑白质病变,临床高度怀疑新生儿线粒体病,建议按照流程积极完善基因检测以确诊。针对危重新生儿,极速基因检测对临床早期诊断及治疗更有意义。
新生儿线粒体病患儿常常处于急症、危重症或濒危状态,预后不佳,治疗策略通常以对症及支持治疗为主,以降低致残率与病死率。
推荐意见8 对所有新生儿线粒体病患儿,推荐基于症状的综合管理方案,可考虑使用鸡尾酒疗法。
推荐说明 对于新生儿线粒体病患儿,临床医生需面对不断出现的各个系统器官功能障碍,因此基于症状的管理目标是延缓疾病进展、降低病死率,为可能的靶向治疗争取时间,包括纠正乳酸性酸中毒、及时治疗感染、补液预防脱水及维持内环境稳定、营养支持、控制癫痫发作等。对于运动障碍、精神发育迟缓和骨关节异常等并发症,应采用物理治疗、体位治疗、矫形辅助和(或)手术矫正。
线粒体鸡尾酒疗法是一种非特异性治疗方法,能最大限度发挥线粒体复合物的残余功能,减轻线粒体中的氧化应激和氧化还原失衡[48, 49],常见成分包括抗氧化剂和氧化应激猝灭剂(如N-乙酰半胱氨酸、α-硫辛酸、泛醌或辅酶Q10)、参与线粒体功能的辅助因子(如各类B族维生素与脂溶性维生素),以及其他营养补充剂(瓜氨酸、左卡尼汀和肌酸)[49, 50, 51]。但目前尚无强有力的证据表明线粒体鸡尾酒疗法对线粒体病有效[52],新生儿使用线粒体鸡尾酒疗法的临床经验及证据不足,可考虑使用左卡尼汀、辅酶Q10、维生素B1、维生素B2、维生素C、维生素E和牛磺酸。现列举鸡尾酒疗法具体剂量,左卡尼丁30~300 mg/(kg·d),辅酶Q10 5~50 mg/(kg·d),维生素B110~20 mg/(kg·d),维生素B2 5~20 mg/(kg·d),维生素C 500 mg/d、维生素E 100~400 IU/d和牛磺酸1~10 g/d。
推荐意见9 对于确诊的新生儿线粒体病患儿,可根据科学证据和理论推理,经家属签署知情同意书、医院相关部门多学科讨论、获医院伦理审批后尝试进行特异性治疗。
推荐说明 对部分发病机制相对明确的线粒体病可尝试进行特异性治疗,包括清除体内有毒物质、酶替代、异基因造血干细胞移植、分子旁路及辅助因子治疗等。N-乙酰半胱氨酸和甲硝唑是乙基丙二酸脑病姑息性治疗的标准方案[53];线粒体神经胃肠道脑病综合征是第一个通过酶替代疗法(红细胞封装重组大肠埃希菌TP)治疗的线粒体病[54],异基因造血干细胞移植可有效降低血液胸腺嘧啶和脱氧尿苷水平,从而改善线粒体神经胃肠道脑病综合征患儿的症状[55, 56, 57];对于酶缺乏引起的线粒体病可使用缺乏的酶产物恢复生化稳态,例如原发性辅酶Q10缺乏症使用辅酶Q10补充剂治疗[57, 58, 59];艾地苯醌可用于预防辅酶Q10缺乏症患儿的视力障碍和促进视力恢复[59, 60, 61];Fujii等[62]研究证实短期内使用丙酮酸钠可改善MELAS综合征患儿的症状;Ohsawa等[63]对10例反复卒中发作的MELAS综合征患儿实施临床试验,发现每日口服大剂量牛磺酸后卒中发作次数减少;大剂量维生素B2可显著改善ACAD9基因变异患儿的乙酰辅酶A脱氢酶活性,缓解脑病、心肌病和肌肉病[64];维生素B1或生物素反应性脑病患儿,服用维生素B1和生物素疗效良好[65];大剂量维生素B1 对丙酮酸脱氢酶复合物缺陷患儿疗效显著[66]。但新生儿线粒体病采用上述靶向治疗的报道极少,将来仍需大量的临床研究。
推荐意见10 对于疑似新生儿线粒体病的患儿,禁用丙戊酸盐、氨基糖苷类抗生素;慎用影响线粒体代谢功能的药物,如推荐说明中的抗癫痫药物、利奈唑胺、丙泊酚、大环内酯类药物、对乙酰氨基酚及阿司匹林。
推荐说明 线粒体病患儿应尽量避免使用一些药物,虽然该类药物在新生儿期使用概率较小,但临床医生有必要知晓:(1)抗癫痫药物:丙戊酸盐有导致肝功能损害和癫痫发作的风险,尤其对于POLG基因变异携带者应避免使用[67];卡马西平、苯妥英和苯巴比妥可导致部分线粒体病患儿癫痫发作加重,托吡酯和唑尼沙胺可能使一些线粒体病患儿出现代谢性酸中毒[65, 66],需在严密观察药物不良反应的情况下使用。(2)抗生素类药物:线粒体对氨基糖苷类抗生素敏感,可能增加线粒体病患儿发生听力损失的风险;利奈唑胺会增加乳酸酸中毒的风险,故疑似新生儿线粒体病患儿应避免使用[68]。(3)丙泊酚有潜在的线粒体毒性,线粒体病患儿发生丙泊酚输注综合征的风险可能增加[69]。
推荐意见11 对于nDNA变异所致的线粒体病家庭,遗传咨询可遵循单基因遗传病相关流程和专家共识,通过产前诊断和胚胎植入前遗传检测实施方法明确;对于mtDNA变异所致的线粒体病家庭,生育风险评估与nDNA变异不同,遗传咨询和产前诊断困难较大。针对以上两组基因遗传变异,均建议检测患儿父母变异携带状态,作为患儿是否发生新发变异的参考。
推荐说明 对于nDNA变异导致的线粒体病,变异的致病性分类、家庭的生育选择与孟德尔遗传病相同,可参考美国医学遗传学与基因组学学会制订的《序列变异解读标准和指南》,针对致病性或可能致病性的变异提出建议,作为指导家庭再生育计划的依据。对于mtDNA变异,需遵循母系遗传规律。由于胎儿变异负荷的潜在不稳定性,缺乏将产前变异负荷与出生后观察联系起来的数据,产前诊断仍面临挑战,特别是对于变异水平与疾病严重程度相关性复杂的情况,需要特别谨慎地进行评估和决策。
推荐意见12 每年检测1次血生化、血气分析、乳酸、维生素D、尿生化、糖化血红蛋白、促肾上腺皮质激素、皮质醇、胰岛素样生长因子、甲状旁腺和甲状腺功能、心电图、心脏彩超和脑电图;2年进行1次血串联质谱、尿气相色谱-质谱、眼科及听力检查。
推荐说明 随访评估包括体重、身长、头围、血压、心率等完整的体格检查和发育评估。定期评估包括每年进行1次体格检查、心脏和神经系统评估。每2年进行1次眼科和听力检查。如有并发症,需特别护理,包括营养支持,例如补充中链甘油三酯;如喂养困难,可经鼻胃管或胃造口进行肠内营养;如呼吸功能不全或睡眠呼吸暂停,需居家无创呼吸支持;如反复感染或严重感染,需进行免疫评估[70]。
线粒体病是由于mtDNA或nDNA致病变异引起的遗传和临床异质性疾病,临床表现各异,优先累及高能量需求的组织和器官,如脑、骨骼肌、眼、心脏、肝脏和肾脏。新生儿线粒体病的症状和体征不典型,易与非线粒体病和其他新生儿并发症相混淆。因此本共识针对新生儿线粒体病的早期识别、实验室及辅助检查进行了说明,并提出了新生儿线粒体病的诊断流程以及目前可行的治疗原则。新生儿线粒体病诊断困难,不能仅仅根据表型诊断,要结合多种实验室检查及基因检测。目前治疗策略有限,仍以对症治疗为主,线粒体病的相关发病机制、诊断及治疗研究仍在不断探索中,故本共识内容将进一步依据相关研究进展进行更新与修订。
本共识在国际实践指南注册与透明化平台完成注册。核心专家组(全体作者)全程参与了共识制订,如实填写利益冲突表,线上会议讨论共识选题及写作纲要,根据条目分工撰写推荐意见和推荐说明。于2024年4月9日形成初稿,经2次线上和线下讨论,形成讨论稿。2024年8月14日分别向全国44名新生儿、神经、代谢内分泌、遗传及罕见病专家发送征求意见稿,所有专家均回复并对共识提出17条修改意见和建议,核心专家组针对所有意见进行多次讨论和修改,形成终稿。
参与本共识审阅的专家(按单位及姓氏汉语拼音排序):重庆市妇幼保健院(苗静琨);重庆医科大学附属儿童医院(宋萃);福建医科大学附属第一医院(陈万金);复旦大学附属儿科医院(李文辉);广东医科大学附属东莞儿童医院(陆小梅);广州医科大学附属妇女儿童医疗中心(韩瑾、张文);海南医科大学第一附属医院(张秋月);暨南大学附属第一医院(宋元宗);南通市妇幼保健院(王学谦);宁波大学附属妇女儿童医院(李海波);青岛大学附属医院(陈志红、姜红);山东大学附属儿童医院(李晓莺);上海交通大学医学院附属仁济医院(张硕);上海交通大学医学院附属上海儿童医学中心(王纪文、王秀敏、郁婷婷);首都医科大学附属北京儿童医院(郝婵娟、桑艳梅);四川省妇幼保健院(汪雪雁);苏州大学附属儿童医院(陈婷、王红英);同济大学附属妇产科医院(张军玉);厦门大学附属妇女儿童医院(郭奇伟);新乡医学院第一附属医院(唐成和);浙江大学医学院附属儿童医院(童凡、吴蔚);郑州大学第三附属医院(张琳琳);中山大学附属第六医院(郝虎)
逯军, 王丹虹, 钟柏茂, 等. 新生儿线粒体病诊断与治疗专家共识(2024)[J]. 中华新生儿科杂志, 2024, 39(11): 647-655. DOI: 10.3760/cma.j.issn.2096-2932.2024.11.002.
所有作者声明无利益冲突

























