肾上腺脑白质营养不良(adrenoleukodystrophy,ALD,OMIM# 300100)是最常见的溶酶体病之一,属于X-连锁遗传病,其致病基因ABCD1发生突变后,其表达的ALD蛋白(ALDP)功能异常,使得极长链脂肪酸(VLCFAs)不能转膜进入细胞溶酶体进行脂肪酸氧化,导致VLCFAs在细胞和体液内异常堆积,细胞和血浆中VLCFAs水平升高,出现弥散性神经脱髓鞘和肾上腺皮质功能不足的临床表现。激素替代和骨髓移植是可能的治疗方法,现重点介绍ALD的遗传病理、诊断与治疗,为临床工作提供帮助。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD,OMIM# 300100)是一种先天性代谢性疾病,也是最常见的溶酶体病之一[1]。ALD属于X-连锁遗传病,其病因是位于X染色体上ATP结合匣D亚组膜1(ATP–binding cassette subfamily D,member 1,ABCD1)基因发生突变后,其表达的ALD蛋白(ALDP)功能异常,使得极长链脂肪酸(VLCFAs)不能转膜进入细胞溶酶体进行脂肪酸氧化,VLCFAs在细胞和体液内异常堆积,特别是脑白质、脊髓、肾上腺及睾丸中,导致细胞和血浆中VLCFA水平升高,出现弥散性神经脱髓鞘和肾上腺皮质功能不足的临床表现。受累病例临床表现差异性大,根据其临床表现的不同分为儿童脑病型(childhood cerebral adrenoleukodystrophy,CCALD)、肾上腺神经脱髓鞘型(adrenomyeloneuropathy,AMN)和单纯肾上腺皮质功能减退型(addison–only)。本病发病率约为1/1.7万,在世界不同地区均有报道,无明显的种族差异,随着新生儿遗传代谢病筛查的普及,实际发病率可能更高[2,3,4]。现着重介绍ALD的遗传病理、诊断及干预新进展,以提高儿科医师对ALD的认识,为临床工作提供帮助。
ABCD1基因位于X染色体长臂2区8带位置,长度为20 000 bp,包含10个外显子,其中可转录DNA长度为3 361 bp,编码区DNA片段长度为2 236 bp,外显子1最长,包含900 bp。迄今,尽管ALD的发病机制未完全阐明,大量证据表明ABCD1基因突变在ALD发病中发挥非常重要的功能。截止到2012年,已经发现ABCD1基因有600种病理多态性[5],其中一半以上是致病性的,突变类型包括错义突变(62%),阅读框移位突变(22%),无义突变(10%),框内缺失和插入(3%),大片段缺失(3%)。最常见的病理性突变是位于5号外显子的c.1415-1416delA G错义突变[6],约占所有族群的8%,已报道的中国人ALD病例中未见热点突变,但常见的突变区域在外显子1和外显子6[7,8,9]。
正常的ALDP位于溶酶体膜上,由745个氨基酸组成,是ATP结合匣(ATP–binding cassette,ABC)蛋白转运子家族成员之一,ALDP和ABC蛋白的功能是负责将VLCFAs从细胞质转运至溶酶体内进行脂肪酸氧化降解过程。
当ABCD1基因突变时,其表达的ALDP减少70%,转运乙酰化的VLCFAs能力降至正常的30%以下,尽管目前患者ALDP下降的原因不十分明确,可能是无义突变导致的,其结果是VLCFAs(C26和C24)在细胞内的聚集,不能被转运至溶酶体内被氧化降解,影响细胞膜的结构、稳定性和功能,造成直接和间接的细胞毒性。表现为肾上腺髓质细胞内促皮质素(ACTH)刺激皮质醇释放能力降低[10],神经细胞氧化磷酸化信号通路下调,过氧化反应加速细胞凋亡发生,出现神经脱髓鞘病变[11]。
尽管研究表明ABCD1基因突变是导致ALD的重要机制,但ALD的临床表现差异性大。即使同胞共患ALD后的表型差异非常大,发病时间可能在儿童期,不少病例会到中年甚至老年才发病,通过单核苷酸多态性(SNP)分析可能会在常染色体上存在一个ABCD1基因的调节基因,也可能存在多个调节因子调控ALD的病理变化,表观遗传及环境因素也与ALD的病理过程相关。血浆中VLCFAs水平和ABCD1基因突变类型不能预测ALD的临床表型轻重。
临床上根据其发病年龄及临床主要表现通常分为3型。
CCALD约占ALD的35%,男性,通常在4~8岁起病,高峰发病年龄为7岁,很少在3岁前发病。临床表现通常表现为行为变化和学习能力下降,多动、注意力下降,常易被误诊为多动症或注意力缺陷,这些症状通常持续数月或更久,并出现更多更严重的神经系统异常,如进行性智力和运动能力倒退、书写困难、失语、阅读困难、视力障碍、平衡力下降。此时行头颅磁共振成像(MRI)会发现脑白质异常改变。个别病例以惊厥为首发症状。病情可在数周内迅速恶化,6个月至2年死亡。同时,绝大多数病例同时有肾上腺皮质功能低下的临床表现。
AMN占40%~45%,典型表现为在20岁后或在中年起病,临床表现为双下肢无力、僵硬,精神异常,性功能障碍(阳痿),这些症状通常缓慢加重或进展停滞,时间跨度会超过10年或更长。40%~45%的AMN伴随神经系统异常MRI表现。其中10%神经系统会进行性加重,导致严重的认知和行为异常,直至完全丧失功能及死亡。70%的病例在出现神经系统病变时合并肾上腺功能皮质功能异常。
单纯肾上腺皮质功能减退型约占10%,起病年龄为2岁至成年期,最常见于7岁时出现肾上腺功能不足,包括难以解释的呕吐、乏力或突发昏迷,皮肤色素加深因人而异出现,通常在肢体暴露及关节部位皮肤色素加深明显。直到有AMN的临床表现,临床上常被诊断为addison病。以上3型中90%的CCALD和70%的AMN均有肾上腺功能异常表现,其母亲携带者通常表型正常。肾上腺功能异常典型的表现为血浆ACTH水平升高,ACTH负荷试验皮质醇升高,无肾上腺抗体。此外,临床上有5%~10%的ALD病例报道,表现为局部脑病型,仅有基因突变,无神经及内分泌功能改变。
有以下临床表现者,临床应考虑为ALD。(1)儿童期起病的男性,表现为注意力缺陷合并进行性智力下降和行为异常、视力下降、语言理解力下降、书写困难等神经系统异常者;(2)年轻及中年起病,进行性步态异常,下肢僵硬、乏力,精神异常、性无能,可合并或不合并肾上腺功能不足、行为及认知异常者;(3)原发性肾上腺皮质功能低下的男性,无论有无神经系统异常者;(4)成年女性,进行性偏瘫、精神异常、下肢感觉异常,有阳性ALD家族史。
85%的病例神经脱髓鞘病变首发于胼胝体压部白质并逐渐扩散到相邻的顶-枕叶白质区域,部分病例神经脱髓鞘初发于胼胝体膝部然后对称性进展至额叶区白质,少数病例是初发于内囊或桥脑的锥体束然后向半圆中心发散。无论其临床表现轻重,头颅MRI平扫或加强检查时,在脑白质脱髓鞘区域T2和压水反转(FLAIR)序列会显现高密度异常信号影,T1序列会显现低密度信号影。头颅MRI检查发现的神经系统异常通常是诊断ALD的首发线索。
脂肪酸定量检测是最重要的辅助检查手段,通过串联质谱分析发现,ALD血浆中VLCFAs水平异常,其中3个指标:C26:0水平、C24:0与C22:0比值、C26:0与C22:0比值异常升高,见表1[12]。
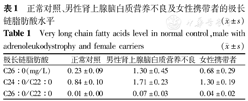
正常对照、男性肾上腺脑白质营养不良及女性携带者的极长链脂肪酸水平( ±s)
±s)
Very long chain fatty acids level in normal control,male with adrenoleukodystrophy and female carriers( ±s)
±s)
正常对照、男性肾上腺脑白质营养不良及女性携带者的极长链脂肪酸水平(±s)
Very long chain fatty acids level in normal control,male with adrenoleukodystrophy and female carriers(±s)
| 极长链脂肪酸 | 正常对照 | 男性肾上腺脑白质营养不良 | 女性携带者 |
|---|---|---|---|
| C26:0(mg/L) | 0.23±0.09 | 1.30±0.45 | 0.68±0.29 |
| C24:0/C22:0 | 0.84±0.10 | 1.71±0.23 | 1.30±0.19 |
| C26:0/C22:0 | 0.01±0.00 | 0.07±0.03 | 0.04±0.02 |
目前已证明,ABCD1基因突变是导致ALD发病的唯一基因。通常采用基因测序进行分析ABCD1有无突变,此项技术可发现99%的男性的ABCD1突变和93%的女性ABCD1携带者,突变类型包括无义突变、错义突变、编码框偏移突变及外显子启动区的突变等,是最经典的分子诊断技术。对于未能发现基因突变者临床有高度怀疑ALD的病例,可以再应用多重连接探针扩增技术(MLPA)、定量PCR及微阵列分析检出ABCD1基因大片段的缺失或重复。
确诊ALD必须进行神经系统的全面评估、头颅MRI检查,肾上腺皮质功能测定、血浆脂肪酸分析及遗传咨询。
根据临床表现及头颅MRI异常表现,再测定VLFA水平可以确诊,有条件者建议行ABCD1基因突变分析。
诊断步骤分为2步。第一,首先确定家族中先证者的基因突变类型;第二,在确诊先证者后,再在家族成员中确定携带者,患者母亲及同胞兄弟姐妹,也包括女性携带者的父亲,无论有无ALD的症状或症状严重程度,均可以通过对测定其家庭成员血浆中VLCFA水平和ABCD1基因测序确定是否携带基因突变,20%的女性携带者血浆中VLCFA水平是正常的。
在确定先证者生化改变及基因突变后,同时证实其母亲是携带者的前提下,在先证者母亲再次妊娠时进行脐带血或羊水生化及基因突变检查,诊断方法同先证者诊断过程。
应注意与多动症、其他肾上腺皮质功能低下、颅内肿瘤、异染性脑白质营养不良、球型细胞脑白质营养不良(Krabbe病)、亚急性硬化性脑炎、多发性硬化、Lyme病、神经脂褐质沉积症少年型进行鉴别。
应注意与多发性硬化、进行性痉挛性截瘫、肌萎缩侧索硬化、维生素B12缺乏症鉴别。
临床表现为肾上腺皮质功能低下的男性,无论有无神经系统异常表现,都要检测血浆中的VLCFA水平,此外需要与Allgrove综合征(无泪、肢体乏力、交感神经系统异常、肾上腺皮质功能低下)相鉴别。
ALD患者及家族成员均应进行遗传咨询。作为X-连锁遗传疾病,95%的ALD病例遗传自母亲携带者,4.1%的ALD病例为新发突变[13]。已诊断ALD,遗传咨询包括肾上腺皮质功能检查,每6个月复查头颅MRI。先证者家族及后代的遗传咨询在确诊后进行。
遗传ABCD1基因突变的风险值分别为:先证者的母亲在证实为携带者后,其下次妊娠时将致病基因下传的概率为50%,生男孩发生ALD的概率为50%,生女孩携带ALD致病基因的概率为50%。如果男性携带致病性ABCD1基因,所生女孩全部为携带者,男孩则正常。
产前诊断适用于在已经诊断为ALD或ALD携带性或有可能是携带者的育龄夫妇中进行。携带者母亲妊娠时,如果不清楚性别,胎儿发生ALD的风险率为25%。产前诊断的时机及步骤为在孕10~12周取绒毛膜细胞或孕15~20周羊水细胞进行基因分析确定有无基因突变。
需要强调的是无论遗传咨询或产前诊断必须在有国家认定资质机构进行。
目前,ALD缺乏特异性治疗方法,干预治疗原则是激素替代改善肾上腺皮质功能,饮食干预降低体内VLCFAs水平,康复训练改善运动功能倒退,早期诊断的CCALD建议行骨髓移植。
当临床出现肾上腺功能不足时,要开始肾上腺糖皮质激素替代治疗,可采用地塞米松、甲泼尼龙,在挽救生命的同时对神经系统无不良影响;随着疾病的进展,可能出现盐皮质激素分泌不足,需要给予补充盐皮质激素。
提倡低脂饮食,国外通过给轻症的CCALD患儿使用Lorerzo油[油酸三甘油酯(glycerol trioleate)与芥子酸三甘油酯(glycerol trierucate) 4:1比例的混合物,2种脂质分别是油酸(oleic acid)与芥子酸(erucic acid)的三酸甘油酯结合型]代替普通的食用油,减缓了其神经系统病变的程度和进展,提示降低C26:0摄入对疾病恢复有帮助[14]。
对肢体功能障碍针对性地进行康复训练,可改善神经系统病变的程度和恶化。心理干预可改善精神及行为异常。


























