
维生素B6相关性癫痫是指癫痫发作不能被抗癫痫药物控制,但可被维生素B6控制或明显改善的一组癫痫相关性疾病,主要包括吡哆醇依赖性癫痫和吡哆醇反应性癫痫,后者又包括维生素B6反应性婴儿痉挛、磷酸吡哆醇(胺)氧化酶(PNPO)缺乏症、高磷酸酶症伴智力落后综合征(即Mabry综合征)等。此组疾病临床表现无特异性,均可表现为新生儿期或婴儿期出现难治性癫痫发作,需与新生儿缺氧缺血性脑病、大田原综合征和非酮症性高甘氨酸血症等疾病鉴别。吡哆醇依赖性癫痫、PNPO缺乏症和Mabry综合征有相对特异的生化标志物,且有明确的致病基因,有助于诊断。推荐对所有早期出现癫痫发作的患儿(包括所有婴儿痉挛患儿),首先试用吡哆醇或磷酸吡哆醛治疗,防止遗漏此组疾病。确诊后,依具体疾病应用维生素B6长期或终生维持治疗。


版权归中华医学会所有。
未经授权,不得转载、摘编本刊文章,不得使用本刊的版式设计。
除非特别声明,本刊刊出的所有文章不代表中华医学会和本刊编委会的观点。
维生素B6作为人体基本的营养物质之一,因其广泛存在于各类食物中并且易于获得,有时被称为"不会缺乏的维生素"。维生素B6由6种同效维生素组成:吡哆醇、吡哆醛、吡哆胺及其经磷酸激酶催化而生成的5′-磷酸盐,即磷酸吡哆醇、磷酸吡哆醛(pyridoxal phosphate,PLP)和磷酸吡哆胺。磷酸吡哆醇、磷酸吡哆胺在肝脏需要通过磷酸吡哆醇(胺)氧化酶[pyridox(am)ine-5′-phosphate oxidase,PNPO]的作用而生成PLP,仅有PLP可通过中枢神经系统细胞膜,发挥转氨、脱羧、调节基因表达等生物活性,维持正常的生理功能[1](图1)。1940年,Spies等[2]首次报道将维生素B6应用于癫痫的治疗。随后,维生素B6被广泛应用于各种癫痫及癫痫综合征。维生素B6相关性癫痫是指癫痫发作不能被抗癫痫药物控制,但可被维生素B6控制或明显改善的一组疾病,主要分为吡哆醇(维生素B6)依赖性癫痫(pyridoxine dependent epilepsy,PDE;OMIM 266100)及吡哆醇反应性癫痫(pyridoxine responsive epilepsy,PRE)2大类,后者又包括维生素B6反应性婴儿痉挛、PNPO缺乏症(OMIM 610090)、高磷酸酶症伴智力落后综合征(hyperphosphatasia mental retardation syndrome,HPMR;OMIM 239300,即Mabry综合征)等。
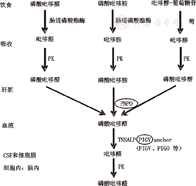
注:PK:磷酸激酶;PNPO:磷酸吡哆醇(胺)氧化酶;TNSALP:组织非特异性碱性磷酸酶;PIGV:磷脂酰肌醇聚糖家族V;anchor:锚;PIGO:磷脂酰肌醇聚糖家族O; CSF:脑脊液 PK:phosphokinase;PNPO:pyridox(am)ine phosphate oxidase;TNSALP:tissue non-specific alkaline phosphatase;PIGV:phosphatidylinositol glycan V;PIGO:phosphatidylinositol glycan O;CSF:cerebrospinal fluid
PDE由Hunt等[3]在1954年首次报道,是婴幼儿期起病的难治性癫痫和癫痫性脑病之一。其特征为大剂量吡哆醇治疗有效,而常规抗癫痫药物无效。2006年,致病基因乙醛脱氢酶7家庭成员A1(aldehyde dehydrogenase 7 family member A1,ALDH7A1)被发现[4],至今,全世界共报道200余例基因确诊病例[5]。关于患病率的报道较少,且不同国家的数据悬殊,介于1︰700 000[6]~1︰20 000[7]。2013年,杨志仙等[8]报道了国内首例PDE患者,我国目前共报道12例病例[9,10],由于病例数少,尚无明确的患病率。
PDE的典型临床表现为新生儿期或婴儿早期即出现难以控制的癫痫发作,发作形式多样,包括局灶性发作、痉挛发作、肌阵挛发作、强直阵挛发作、失张力发作,甚至癫痫持续状态等[5,11,12]。发作对抗癫痫药物耐药,可通过大剂量吡哆醇完全控制且需终生维持治疗,一旦停用吡哆醇,癫痫发作会在1~51 d复发[11]。多数患者单次静脉给予50~100 mg吡哆醇可在数分钟内终止发作,少数需重复给药,也有小剂量用药即可完全终止发作的报道[13]。少数病例初次应用吡哆醇治疗时,随惊厥停止可出现短暂的昏迷、肌张力减低、呼吸不规则等,因此,有条件者应在脑电图(EEG)和呼吸监护下给药[13]。此外,约1/3的患儿临床表现不典型,主要包括癫痫发作出现晚,最晚可晚至3岁[5];发作最初可通过抗癫痫药物控制或最初应用吡哆醇无效;停用吡哆醇后癫痫复发间隔时间长,最长可达5.5个月[14]。部分患儿母亲孕期曾觉察有异常胎动,提示存在胎儿期癫痫发作的可能[15],出生时可出现Apgar评分减低和低脐带血氧等窒息表现,出生后常伴明显的烦躁、入睡困难和呕吐等脑病表现,易被误诊为缺氧缺血性脑病,应注意鉴别。
本病EEG缺乏特异性,常见的发作间期EEG表现为背景活动异常伴各种阵发性异常,包括局灶性、多灶性或广泛性癫痫样放电、不连续图形,如暴发-抑制、高度失律等[13];发作期EEG因发作类型不同而异。少数患者吡哆醇治疗前后EEG均正常;头颅磁共振成像(MRI)可正常,或出现多种非特异性异常,包括胼胝体发育不良、脑室扩大、脑萎缩、内侧颞叶硬化、皮质发育不良等[5]。
2006年ALDH7A1基因被确定为PDE的致病基因[4],为常染色体隐性遗传。ALDH7A1基因定位于5q31,全长53 550 bp。目前,全世界共报道80余种不同突变位点,包括错义、无义、缺失、插入和剪切位点突变等,其中错义突变约占60%。突变c.1279 G>C(p.Glu427Gln)(NM 001182.4)在白种人中出现率高,约占33%,考虑可能是白种人的热点突变[16,17];在我国患儿中,第11内含子IVS11+1G>A突变出现率达31.25%,可能为我国PDE的热点突变[9]。既往研究中,多数患儿检出ALDH7A1基因纯合突变或复合杂合突变,但有6例仅检出1个突变位点[11,16,17,18],近期经微阵列比较基因组杂交技术(array-CGH)检测,5例检出了长度介于1 700~70 000 bp的片段缺失[19]。因此,对于临床诊断明确但未检出基因突变或仅检出1个突变位点者,应进一步行array-CGH或多重连接探针扩增技术(MLPA)检测,防止遗漏拷贝数变异。
ALDH7A1基因编码α-氨基己二酸半醛(α-ami-noadipic semialdehyde,α-AASA)脱氢酶,参与体内赖氨酸的分解代谢,该基因突变会引起α-AASA累积,后者在体内与Δ1-四氢吡啶-6-羧酸(delta-1 piperideine-6-carboxylic,P6C)处于自发平衡状态,导致P6C继发性累积,并进一步引起体内哌啶酸(pipecolic acid,PA)累积[4,17]。因此,PDE患者血液、尿液、脑脊液中α-AASA、P6C及PA浓度升高,三者均可作为诊断PDE的生化标志物[5]。其中,α-AASA和P6C具有较高的特异性,但稳定性差,在室温下极易降解[20],难以精确定量;PA稳定性好、灵敏度高,但特异性相对较低,除PDE外,尚可见于过氧化物酶功能障碍、高赖氨酸血症、脯氨酸代谢缺陷、肝功能异常等疾病[5]。PDE患儿个体间生化标志物水平差异较大,可能与基因突变类型、年龄、吡哆醇治疗及营养性赖氨酸摄入量等有关[13]。长期应用吡哆醇治疗后各生化标志物可明显降低甚至恢复正常[17]。
PDE患者需终生补充吡哆醇,目前长期维持治疗的剂量尚不明确。婴儿一般推荐剂量为15~30 mg/(kg·d),新生儿可高达200 mg/d,成人可高达500 mg/d[5],上述剂量长期治疗的安全性已经得到证实。当遇到急性发热性疾病时,可通过增加吡哆醇剂量来预防或控制暴发性的惊厥发作[13]。由于大剂量维生素B6治疗可能引起少见的肝功能障碍、感觉或运动周围神经病等不良反应,且多数可逆,治疗过程中推荐定期复查肝功能,并定期行头颅MRI,以监测脑的髓鞘化变化[13]。PLP是吡哆醇的活性形式,也可作为PDE的一线用药,可予30 mg/(kg·d),分3次口服[13]。另有部分患者对亚叶酸治疗有效,吡哆醇治疗过程中添加3~5 mg/(kg·d)亚叶酸治疗可能有一定益处[21]。此外,限制赖氨酸摄入理论上也是一种治疗方法,但临床疗效尚需进一步研究证实[5]。
由于PDE的常染色体隐性遗传特性,患儿父母再生育仍有25%的患病风险。对于产前诊断确诊者若不终止妊娠,产前补充吡哆醇可能会阻止胎儿宫内惊厥发作,并改善神经发育。自妊娠早期起给予孕妇100 mg/d的吡哆醇是安全的,对胎儿无任何不良反应,同时还可以治疗妊娠呕吐[6]。
PRE由Hansson和Hagberg[24]于1968年首先提出,指癫痫发作可被维生素B6单药控制,或在已有抗癫痫药物不能控制发作的基础上,加用维生素B6后发作控制达1个月以上[25]。其涉及各种癫痫发作类型及多种癫痫综合征,如婴儿痉挛、PNPO缺乏症、Mabry综合征、低磷脂酶症和高脯氨酸血症Ⅱ型、Lennox-Gastaut综合征等[5],其中以婴儿痉挛最多见[25,26]。
1977年,Ohtahara等[27]首次提出将大剂量维生素B6用于婴儿痉挛的治疗。研究发现,10%~30%的婴儿痉挛患儿对大剂量维生素B6治疗反应良好[28]。在日本和欧洲,大剂量维生素B6已成为治疗婴儿痉挛的首选药物[24,26]。维生素B6对特发性/隐源性和症状性婴儿痉挛均可能有效,对前者有效率更高;在症状性婴儿痉挛中,有报道对病因为结节性硬化症者更为有效[29]。
维生素B6反应性婴儿痉挛的临床表现无特异性,均为婴儿期起病,以痉挛发作为主,发作间期EEG表现为高度失律图形[30]。尽管有报道维生素B6对少数Lennox-Gastaut综合征患儿有效[31],但多数情况下,维生素B6仅在婴儿痉挛阶段有效,一旦复发,转变为局灶性癫痫或Lennox-Gastaut综合征等,则难以通过维生素B6单药或联合其他抗癫痫药物控制[25]。维生素B6反应性婴儿痉挛对维生素B6的反应十分迅速,80%的痉挛发作在用药1周内明显减少[31],多在2周[28]或20 d[25]内完全控制,随发作控制EEG迅速改善。本课题组此前的研究中,90%的患儿在用药1周内发作完全控制,但仍有1例在用药1.5个月后发作控制[30]。因此,对于维生素B6用药之初癫痫发作不能被完全控制者,不能排除维生素B6反应性癫痫的可能,应根据其对维生素B6的反应考虑适当提高剂量或延长治疗时间,以明确诊断和治疗效果。
维生素B6反应性婴儿痉挛的机制尚不清楚,推测可能存在年龄相关或维生素B6依赖的神经递质功能不成熟相关的酶活性异常[31]。有研究发现婴儿痉挛患儿脑脊液中抑制性神经递质γ-氨基丁酸(GABA)浓度明显低于健康人群[32];且经大剂量维生素B6治疗后可恢复正常[33],因此,推测维生素B6可能通过影响GABA的浓度达到控制癫痫发作的作用[28]。随着研究进展,越来越多的证据表明婴儿痉挛可能由脑发育的关键基因调控通路紊乱引起:大脑背腹侧的GABA能基因调控网络和突触相关基因表达异常[34]。目前,已报道10种与婴儿痉挛相关的致病基因,包括ARX、CDKL5、FOXG1、GRIN1、GRIN2A、MAGI2、MEF2C、SLC25A22、SPTAN1和STXBP1[35]。因此,维生素B6反应性婴儿痉挛亦可能由基因突变导致,但在本课题组报道的10例中均未检测到上述基因突变[30],未来需通过全外显子测序等方法进一步寻找可能的致病基因。
目前维生素B6治疗婴儿痉挛的剂量和疗程尚无统一标准,建议以不低于10 mg/(kg·d)的剂量开始应用[30],可用至100~200 mg/d或20~30 mg/(kg·d),分3次口服,出现恶心、呕吐等不良反应时,可分为更多次口服;若1周后疗效不显著,可将剂量加至300~400 mg/d或40~50 mg/(kg·d),并注意观察发作频率的变化及药物耐受性[31]。大剂量吡哆醇长期维持治疗的不良反应少见,多出现于剂量超过400 mg/d的患儿,且多数可逆,减量或停药后常可迅速消失[29],包括情绪淡漠、食欲减退、恶心、呕吐、腹胀、腹泻、便秘、出血性胃炎、肝功能障碍、感觉或运动周围神经病及横纹肌溶解等[33,36,37],其中以胃肠道症状最为常见[28]。
与PDE不同,维生素B6反应性婴儿痉挛治疗后可停药。已报道50.0%隐源性及23.5%症状性患儿停药后未复发,EEG亦未再出现发作间期放电[31]。Ohtsuka等[25]研究中,痉挛发作控制后EEG恢复并持续正常者均未复发,EEG持续异常者50%复发;本课题组此前的研究中,7例EEG持续正常者均未复发,2例EEG持续异常者均因减、停维生素B6复发[30]。因此,EEG在指导治疗、预测复发等方面发挥重要作用,应定期复查。发作控制且EEG恢复正常后,可考虑减停药物。Ohtsuka等[25]推荐自开始减量至完全停药平均需要2年。目前,停药的具体时机尚无定论。有研究认为发作控制且EEG恢复正常者1岁后即可停药[25];而Ohtahara等[31]认为发作控制后EEG持续正常超过2年者,停药后不复发的可能性大。此外,考虑到维生素B6在脑功能方面的重要作用,即使发作控制且EEG恢复正常后,长期维持口服小剂量维生素B6也可作为一种治疗方案[31]。
婴儿痉挛预后较差,多数患儿会遗留不同程度的智能缺陷和运动发育迟滞,仅有约10.7%的患儿智力运动发育正常[24]。而本课题组报道的一组维生素B6反应性婴儿痉挛患儿中,50%的患儿智力运动发育正常[30],显著优于总体水平,提示对维生素B6治疗有效者预后佳,且治疗的早晚与预后密切相关。
综上,推荐对所有婴儿痉挛患儿均应尝试大剂量维生素B6治疗[30,31]:(1)特发性/隐源性或症状性患儿,均有可能通过维生素B6单药控制;(2)维生素B6的不良反应较促皮质素(ACTH)或其他抗癫痫药物少;(3)维生素B6反应性婴儿痉挛患儿的痉挛发作易控制,且智力运动发育多数较好;(4)短期内即可评估维生素B6的疗效,不会延误非维生素B6反应性婴儿痉挛患儿的治疗。
PNPO缺乏症由Kuo和Wang[38]在2002年首次提出,特征为新生儿期即出现严重的癫痫性脑病,癫痫发作对抗癫痫药物无反应,吡哆醇治疗无效或仅有部分疗效[39],发作多可被PLP单药控制,PLP撤药后癫痫发作反复[40]。因此,也曾被称为PLP依赖性癫痫[41]或PLP反应性癫痫[42] (图1)。2005年,PNPO缺乏症的致病基因PNPO被确定[40],明确其常染色体隐性遗传特性。此病较为罕见,目前全世界报道不超过50例[43],国内仅报道2例[44]。
癫痫发作为PNPO缺乏症的主要临床表现之一,多于出生后短时间内出现,既往报道病例中61%出现于出生24 h内,96%出现于出生1个月内[45]。癫痫发作形式多样,以多灶或全面性肌阵挛发作为主(61%),且多不能被常规抗癫痫药物或吡哆醇控制[42,45]。超过半数(57%)的患儿发作间期EEG显示暴发-抑制图形,另有20%显示异常的不连续图形[45]。早产史较常见,出现于61%的患儿中;另有11%的患儿孕期有明显的异常胎动[45]。头颅MRI可正常或为非特异性异常[39]。随着越来越多基因确诊病例被报道,PNPO缺乏症的表现谱逐渐扩展,包括:新生儿早期起病,癫痫发作仅对PLP有反应;婴儿痉挛起病,癫痫发作仅对PLP有反应;3个月内起病,癫痫发作对吡哆醇有反应,对PLP有或无反应[39]。既往报道病例中,80%的患儿曾应用吡哆醇,其中46%有明确的临床反应;52%的患儿曾应用PLP治疗,其中75%临床反应好。研究指出,之所以部分患儿对吡哆醇有反应,可能与其所携带的基因突变对PNPO蛋白构象影响较小,酶功能未完全丧失有关,此时,若大量补充吡哆醇会使体内PNPO的底物(即磷酸吡哆醇)水平升高,从而转变为PLP发挥作用[46]。目前,c.279_290del、p.R141C及p.R225H等突变被认为与吡哆醇反应性相关[47]。然而,4例吡哆醇治疗有效者换用PLP后反应差,甚至症状加重,分别出现癫痫持续状态(2例)、发作复发(1例)或发作频率增加(1例)[45],这可能因为PLP对PNPO的活性有很强的抑制作用,当突然应用大剂量PLP时,原本残留的部分酶活性被完全抑制,致使症状加重[47]。
维生素B6经肠道被动吸收后,首先在肝内转化为磷酸化衍生物,其中磷酸吡哆醇和磷酸吡哆胺在PNPO的作用下氧化生成PLP,并进入血液循环。PLP是维生素B6的唯一活性形式,在血液中与血清清蛋白结合,在组织非特异性碱性磷酸酶(tissue non specific alkaline phosphatase,TNSALP)的作用下水解为吡哆醛,通过血脑屏障进入脑及其他组织,并进一步以PLP的形式参与体内140余种酶促反应[48],包括氨基酸、糖原的代谢及核酸、血红蛋白、鞘磷脂、鞘脂和神经递质(血清素、多巴胺、去甲肾上腺素、GABA)等物质的合成等[49],维生素B6缺乏可出现癫痫、肝大、贫血、低血糖、氨基酸代谢紊乱等相关临床表现[50]。
PNPO基因定位于17q21.2,全长约7 500 bp,包含7个外显子,共编码261个氨基酸。目前,已有超过24种不同的致病位点被报道,以错义突变和无义突变最常见[42]。PNPO基因突变导致PNPO缺乏,导致磷酸吡哆醇和磷酸吡哆胺在肝脏中不能转变为PLP而进入血液,并最终导致体内特别是脑内PLP生成不足(图1),引起多种物质代谢障碍:(1)PLP作为谷氨酸脱羧酶的辅酶,参与抑制性神经递质GABA的合成,PLP缺乏使GABA合成明显减少,引起新生儿期严重的癫痫性脑病;(2)PLP缺乏引起多种PLP依赖性酶的继发性功能障碍,导致血、尿、脑脊液中的氨基酸、神经递质代谢异常,如甘氨酸裂解酶和苏氨酸脱水酶活性降低,导致脑脊液中甘氨酸、苏氨酸水平升高;芳香族氨基酸脱羧酶功能障碍引起脑脊液中高香草酸(HVA)、5-羟吲哚乙酸(5-HIAA)水平降低,进一步引起尿中香草酸(VLA)及脑脊液中3-甲氧基酪氨酸(3-MT)水平升高;δ-鸟氨酸转氨酶活性降低,引起血浆和脑脊液中精氨酸水平降低等[49]。上述代谢产物可作为PNPO缺乏症的生化标志物,但特异性差,且在少数患儿体内可正常,甚至可与上述改变完全相反,如HVA、5-HIAA在部分患儿体内水平可升高[45]。因此,上述生化标志物对诊断有提示性意义,但并不能明确或排除诊断,最终仍需基因分析确诊。
PNPO缺乏症患儿需终生补充PLP。目前长期治疗的剂量尚无明确建议,主要依据患儿对药物的反应进行调整,多介于30~60 mg/(kg·d),分3、4次口服[48]。患感染性疾病期间可将PLP暂时加量以预防或控制发作[48]。与PDE相似,PNPO缺乏症患儿初次应用PLP后也可能出现严重肌张力减低、呼吸暂停等,同期EEG示脑电活动被严重抑制[40]。因此,有条件者初始治疗应在EEG和呼吸监护下进行,及时观察治疗反应及可能出现的呼吸暂停。此外,有研究报道长期应用PLP可能引起肝功能异常,甚至出现肝硬化[48],可能与PLP剂量过高或片剂溶于液体的过程中产生毒性降解产物有关[48],因此,推荐PLP应直接以片剂形式口服,或溶解后立即服用。另有研究认为肝功能异常可能为PNPO缺乏症的一种少见的临床表现[43]。尽管机制尚不明确,但PLP治疗过程中对肝功能进行定期监测是非常必要的。
PNPO缺乏症患儿预后差异较大,多数发作可控制,智力运动发育可正常(63%)或出现落后(37%)[45]。但少数患儿应用吡哆醇和PLP均不能完全控制发作,考虑可能因其延迟治疗时间过长导致继发性难治性癫痫,因此,早诊断、早治疗有助于获得良好预后。若治疗不及时,患儿多于出生2~24周死亡,即便存活,也会遗留严重的神经系统后遗症[49]。此外,基因型不同也是影响预后的重要因素[51]。
HPMR由Mabry等[53]于1970年首次报道,自2010年起被广泛称为Mabry综合征[54],是一种少见的常染色体隐性遗传病。2010年,PIGV基因被确定为Mabry综合征的首个致病基因[55];随后,PIGO、PGAP2、PGAP3、PIGW、PIGL等致病基因相继被确定[56,57,58,59,60]。目前,全世界仅报道30余例基因确诊病例,其中PIGV基因突变最常见,占半数以上(约20例)[55,61,62,63];另报道PIGO基因突变6例[56,64,65],PGAP2基因突变2例[57],PGAP3基因突变5例[58],PIGW、PIGL基因突变各1例[59,60]。本课题组于2016年首次报道了国内2例Mabry综合征病例,分别携带PIGV、PIGO基因复合杂合突变[64]。
持续性血清碱性磷酸酶(ALP)升高、发育落后和癫痫是Mabry综合征的3个主要临床症状[54]。其中,ALP持续性升高见于所有的Mabry综合征患者,是其特征性表现,可将其与其他症状相似的疾病区分开来,如Coffin-Siris综合征、Cornelia de Lange综合征等[54]。ALP在不同病例间升高程度差异较大,可为正常范围高限的1.5~17.2倍[64],可能与不同致病基因、同一致病基因不同突变位点相关。中-重度智力运动发育落后也是所有Mabry综合征患儿的共同症状。68%的患儿肌张力低;多数患儿不会说话,少数可说简单词语;超过半数的患儿可独走或扶走,但走路年龄明显延迟[55,61,62,63]。有研究认为携带PIGV基因突变的患儿生长发育指标(如头围)多正常或高于正常,而携带PIGO基因突变者常落后于正常水平[56],但在国内2例病例中并未得到体现[64],未来需通过更多病例加以论证。多数患儿病程中会出现癫痫发作,发作形式多样,以肌阵挛发作、强直-阵挛发作为主[55,61,62,63];首次发作年龄不一,早至新生儿期,晚至5岁以后均有可能。癫痫发作对抗癫痫药物的反应差异较大,多数患者可经单药或联合用药控制,常用丙戊酸、氯硝西泮等[62];但部分患者经多种抗癫痫药物治疗无效或仅有部分疗效[61]。Thompson等[66]首次提出吡哆醇可缓解高磷酸酶症患者的神经代谢异常;2013年Kuki等[67]首次报道1例Mabry综合征患者应用吡哆醇治疗后癫痫发作完全控制,且EEG明显改善,提示Mabry综合征可能对吡哆醇有反应,但由于目前病例数少,相关研究不足,未来需进一步明确吡哆醇的作用,可能为此综合征的治疗提供新的治疗方法,改善预后。
除上述3种主要临床表现外,面部畸形、末节指(趾)骨发育不良对此综合征的诊断也有一定的提示作用[64]。其中,特征性的面部畸形出现于绝大多数患儿中,主要表现为眼距宽、睑裂长、鼻梁宽平、鼻尖圆钝、上唇呈帐篷样支起,部分患儿尚可出现上颚高拱、唇/腭裂等[59,60,64]。末节指骨发育不良出现于约87%的患儿中,主要表现为末节指(趾)骨短,伴或不伴指(趾)甲发育不良和指(趾)弯曲[64]。但有3例携带PIGO基因复合杂合突变的患儿未出现上述2种症状[64],可能与不同突变导致蛋白功能改变不同有关,但是否仅出现于PIGO基因突变者尚不明确。面部畸形和末节指骨发育不良并非Mabry综合征诊断所必须,最终均应通过基因分析确诊。
此外,其他多种器官异常在Mabry综合征患儿中也较常见,如先天性巨结肠、肾脏/输尿管/膀胱异常(先天性肾积水、膀胱输尿管反流)、肛门直肠畸形(肛门狭窄/闭锁、直肠前庭瘘)、听力损害、心脏缺陷(房/室间隔缺损、肺动脉狭窄、法洛四联症)、漏斗胸等[64]。
自2010年,PIGV、PIGO、PGAP2、PGAP3、PIGW、PIGL等相继被确定为Mabry综合征的致病基因,均参与糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)的生物合成与重塑过程。其中,PIGV作为最主要的致病基因,出现于半数以上的患者中,共发现10余种不同的突变位点,以错义突变为主,c.1022C>A (p.Ala341Glu)出现率约为72.7%,考虑可能为热点突变[64]。
GPI作为结构复杂的糖脂类化合物在真核生物中广泛表达。蛋白质的C-末端以共价键形式与GPI相连是一种常见的翻译后修饰,GPI修饰的蛋白质可以通过GPI锚定在细胞膜表面。GPI锚及其锚定蛋白在众多生物学过程中发挥重要作用。GPI锚合成后与内质网中相应的蛋白质连接形成GPI锚定蛋白(GPI anchored protein,GPI-APs),GPI-APs转运至高尔基体,经脂肪酸重塑后进一步转运至细胞膜,脂肪酸重塑过程对GPI-APs的成熟和与细胞膜上脂筏的连接至关重要[68]。GPI锚的生物合成和重塑过程中涉及20余种不同的基因,包括与Mabry综合征相关的致病基因[55,56,57,58,59,60]。其中,PIGW、PIGL基因参与GPI锚合成的早期阶段;PIGV、PIGO基因参与GPI锚合成的晚期阶段;PGAP2、PGAP3参与GPI锚在高尔基体内的脂肪酸重塑过程。有研究认为,GPI锚合成的晚期阶段受损会产生连接有甘露糖残基的不完整的GPI锚,而GPI转酰胺酶有识别甘露糖残基的亚基,可被上述不完整的GPI锚激活,使GPI-APs的C-末端疏水信号肽裂解[69];GPI重塑过程受损会导致GPI-APs更容易被裂解[58]。因此,上述过程中相关基因突变均会使锚定于GPI的ALP减少,而分泌到细胞外间隙的ALP增多,导致血清ALP水平升高。而GPI锚合成的早期阶段受损会产生不含甘露糖残基的不完整GPI锚,因GPI转酰胺酶不能被激活,并不会引起血清ALP水平增高[69]。但随着致病基因PIGW、PIGL被发现,证实GPI锚合成的早期阶段受损也会引起ALP升高。因此,Mabry患者出现高磷脂酶血症的机制尚待进一步研究。基因突变导致GPI的生物合成与重塑过程受到影响,进而影响到TNSALP的活性,导致PLP在血液中脱磷酸障碍,而不能通过血脑屏障进入脑内并再次转变为有活性的PLP(图1)。
Mabry综合征目前尚无明确的治疗方法,对于出现癫痫发作的患者给予抗癫痫药物或吡哆醇进行对症治疗。患者均遗留中-重度智力运动发育落后。
综上,维生素B6相关性癫痫是指癫痫发作对抗癫痫药物无反应,可通过吡哆醇/PLP单药控制或明显改善的一组疾病。其中,PDE与PNPO缺乏症均可在宫内即出现惊厥发作,出生时可能出现Apgar评分减低、代谢性酸中毒等,需与缺氧缺血性脑病相鉴别;PDE、PNPO缺乏症及Mabry综合征均可在新生儿期或婴儿早期即出现难治性癫痫发作,EEG无特异性,可能为暴发-抑制图形,需与大田原综合征和非酮症性高甘氨酸血症等相鉴别[70]。再者,上述维生素B6相关性癫痫各疾病间也应注意相互鉴别:PDE与PNPO缺乏症临床表现极相似,但两者均有相对特异的生化标志物,可提示诊断;Mabry综合征有特征性的面部、手/足畸形和血清ALP持续性异常升高;三者均为常染色体隐性遗传病,有明确的致病基因,最终可通过基因检测确诊。此外,有研究报道其他可能对维生素B6治疗有反应的疾病,如低磷酸酯酶症(TNSALP缺乏所致)、高脯氨酸血症Ⅱ型(Δ1-吡咯啉-5-羧酸缺乏所致)等[5],但与维生素B6的相关性尚需进一步研究证实。

























